2. Library Cloning
Upon receiving the oligo pool from TWIST (which typically takes ~1-2 weeks) or IDT, it is necessary to amplify the oligo pool with a small number of PCR cycles to increase the amount of DNA.
In first part of the protocol, we discussed the importance of pooling small groups of mutated sequences together; typically 3-5 genes for each primer pair. This grouping saves time in this part of the protocol, as it eliminates the need to perform a separate PCR reaction for each gene (and its 1200-1500 mutated versions).
The TWIST website includes instructions on oligo pool amplification. We recommend that you familiarize yourself with that page before proceeding.
An initial amplification of the aliquoted oligo library ensures that we have ample DNA for cloning of the library into plasmids, followed by transformation into E. coli or another organism of interest. This part of the protocol assumes that you have divided the oligo library into smaller aliquots, at a concentration of 10 nanograms per microliter. For long-term storage, these DNA libraries should kept at -80C.
When you are ready to work with the DNA, follow the TWIST guidelines to amplify the oligo library.
Remove an aliquot (we recommend 5 microliters per aliquot) of the oligo pool, and set up one 50 microliter PCR reaction per primer pair. If you have 5 primer pairs (corresponding to up to 25 gene libraries), this would be 5 PCR reactions.
For each PCR reaction:
| reagent | concentration | volume (microliters) |
|---|---|---|
| DNA (oligo pool) | 10ng/microliter | 1 |
| Q5 polymerase mix | 2x stock | 25 |
| Primer Fwd | 10 micromolar | 2.5 |
| Primer Rev | 10 micromolar | 2.5 |
| Water | N/A | 19 |
Mix these reagents carefully, while keeping everything on ice throughout the experiment. The 'Primer Fwd' and 'Primer Rev' will differ based on the 20-nucleotide sequences flanking each group within the oligo pool (e.g. Kosuri-101 is a primer set that is distinct from Kosuri-102. Most of these primer pairs are already available in the Phillips laboratory, and are stored in the -20°C freezer).
Use the following thermocycler settings with 12 cycles.
| cycles | temperature | time |
|---|---|---|
| 1 | 98°C | 30 seconds |
| 12 | 98°C | 10 seconds |
| 12 | 64°C (anneal) | 30 seconds |
| 12 | 72°C (extend) | 30 seconds |
| 1 | 72°C | 120 seconds |
| Hold | 4°C | ∞ |
For more information on primer annealing temperatures (and a very user-friendly annealing temperature calculator), visit the NEB website's Tm calculator. For more information on the thermocycler conditions for Q5 polymerase, see the NEB website.
Given the low concentration of DNA involved, even after 12 cycles of amplification, loss of material is a major concern at this stage. Therefore, we recommend that you use column-based DNA purification methods to "clean" the amplified DNA. The Zymo DNA Clean & Concentrator-5 works well for this purpose.
After performing the PCR purification according to manufacturer's protocol, NanoDrop the eluted DNA and record the concentration and purity. Store away DNA at 4°C (if you will use in the next 24 hours) or at -20°C for longer term storage.
Recall that, when ordering the oligo pool, promoters and their mutated versions were clustered into different "primer sets". When ordering 25 mutated promoters, therefore, there should be roughly 5 "groups" of genes with different primer sets. Each gene "group" must be amplified individually, using forward and reverse primers that correspond to the first 20, and last 20, nucleotides of the oligo pool.
The initial amplification step need not add barcodes. It merely serves to increase the amount of DNA for each group of mutated promoters.
After initial amplification, it is time to add barcodes to each of the gene groups. Barcoding DNA, as a process, involves adding random, unique, 20-nucleotide "barcodes" to a DNA sequence of interest. Barcodes assist in downstream sequencing, as each unique barcode is associated with one, and only one, mutated promoter sequence. By "mapping" promoters to a unique barcode, and counting the relative frequency of each barcode via next-generation sequencing (NGS), gene expression can be computed for each mutated promoter.
Genetic barcodes can be added to the oligo pool via PCR, using primers with randomly-synthesized "overhangs". During extension of DNA, these random synthesis regions of the primer are incorporated into the amplified DNA. Adding barcodes via PCR is a biased process, however, wherein some barcodes are incorporated with a higher fidelity than others. Bias is especially pronounced at higher amplification cycles, when more DNA is present in a PCR reaction. Therefore, adding barcodes should be done with care to minimize bias wherever possible.
An important step to minimize bias in barcoding DNA involves performing quantitative PCR (or qPCR) to determine the "optimal" number of cycles to amplify DNA without saturating a PCR reaction mixture. Specifically, we wish to determine the cycle number at which DNA is in exponential amplification, but has not saturated. We then use that number of cycles to perform the "real" barcoding PCR reactions. qPCR also serves as an important control because it ensures that the DNA for each group (with orthogonal primer pairs) is being amplified at relatively even levels.
There are many qPCR reaction mixtures available, which contain all of the salts, buffers, and polymerase necessary for quantitative amplification. These mixtures also contain dyes, which the qPCR machine uses to "read out" the amount of DNA present in a tube after each cycle of amplification.
In the past, our lab has used PerfeCTA SYBR Green SuperMix, Low ROX for qPCR. We used this simply because it was available in lab already, and it worked. Other qPCR reagents, such as the NEB Luna qPCR kit, may also work for this purpose.
When performing qPCR, you should use the actual primers that you will use for barcoding the oligo pools. We discuss the sequence of these "barcoding primers" in the next step of this protocol, after first outlining the qPCR process.
Our lab has an Applied Biosystems qPCR machine with MxPro software installed. To perform qPCR, set up 50 microliter reaction volumes, and also prepare a "blank" control sample (no template DNA added). Mix the following reagents on ice, preparing two reactions PER gene "set", one with 1ng of DNA, and the other with 10ng of DNA. Note that the DNA template, in the reagent list below, is the PCR product which was purified from the previous step in this protocol.
| reagent | volume (microliters) | concentration |
|---|---|---|
| PerfeCTa SYBR Green SuperMix, Low ROX (2X) | 25 | N/A |
| Forward primer | 2.5 | 10 micromolar |
| Reverse primer | 2.5 | 10 micromolar |
| Nuclease-free water | 19 | N/A |
| DNA template | 1 | 10 OR 1 ng/microliter |
The total volume for each reaction, again, is 50 microliters. There should be 2 reactions per primer pair (one with 1ng of DNA and the other with 10ng of DNA used as template). There should also be a reaction in which no DNA was added at all for each primer pair. Replace the DNA template by adding an additional microliter of water.
Set up these qPCR reactions on ice and then load them into the qPCR machine. Specify the wells and reference dye in the MxPro software. The QuantaBio qPCR kit contains a reaction buffer with magnesium chloride, dNTPs (dATP, dCTP, dGTP, dTTP), AccuStart Taq DNA Polymerase, SYBR Green I dye, and a ROX Reference Dye. Input the dye information as necessary in the MxPro software, and then run with the following thermocycler settings (NOTE: AccuStart Taq will demand different settings than, say, the polymerase in the Luna qPCR kit. In an ideal world, you would use the same polymerase for both the qPCR and barcoding PCR reactions, as there are deviations in polymerase behavior (different annealing temperatures, different thermocycler settings, and wildly different fidelities). Check the specific thermocycler settings for each polymerase that you wish to use.
| cycles | temperature | time |
|---|---|---|
| 1 | 94°C | 120 seconds |
| 30 | 94°C | 20 seconds |
| 30 | (anneal temp.) | 30 seconds |
| 30 | 72°C (extend) | 20 seconds |
| HOLD | 4 °C | ∞ |
The annealing temperature should be determined (using NEB's calculator) based on the primers to be used for barcoding. Only those nucleotides in the primer that bind to the template DNA should be used to determine the annealing temperature. Do not include nucleotides which reside in the overhang sequence.
Once the qPCR has finished running, check the qPCR curves, which should resemble the curves shown in the image below, which was taken from ThermoFisher's resource on qPCR.
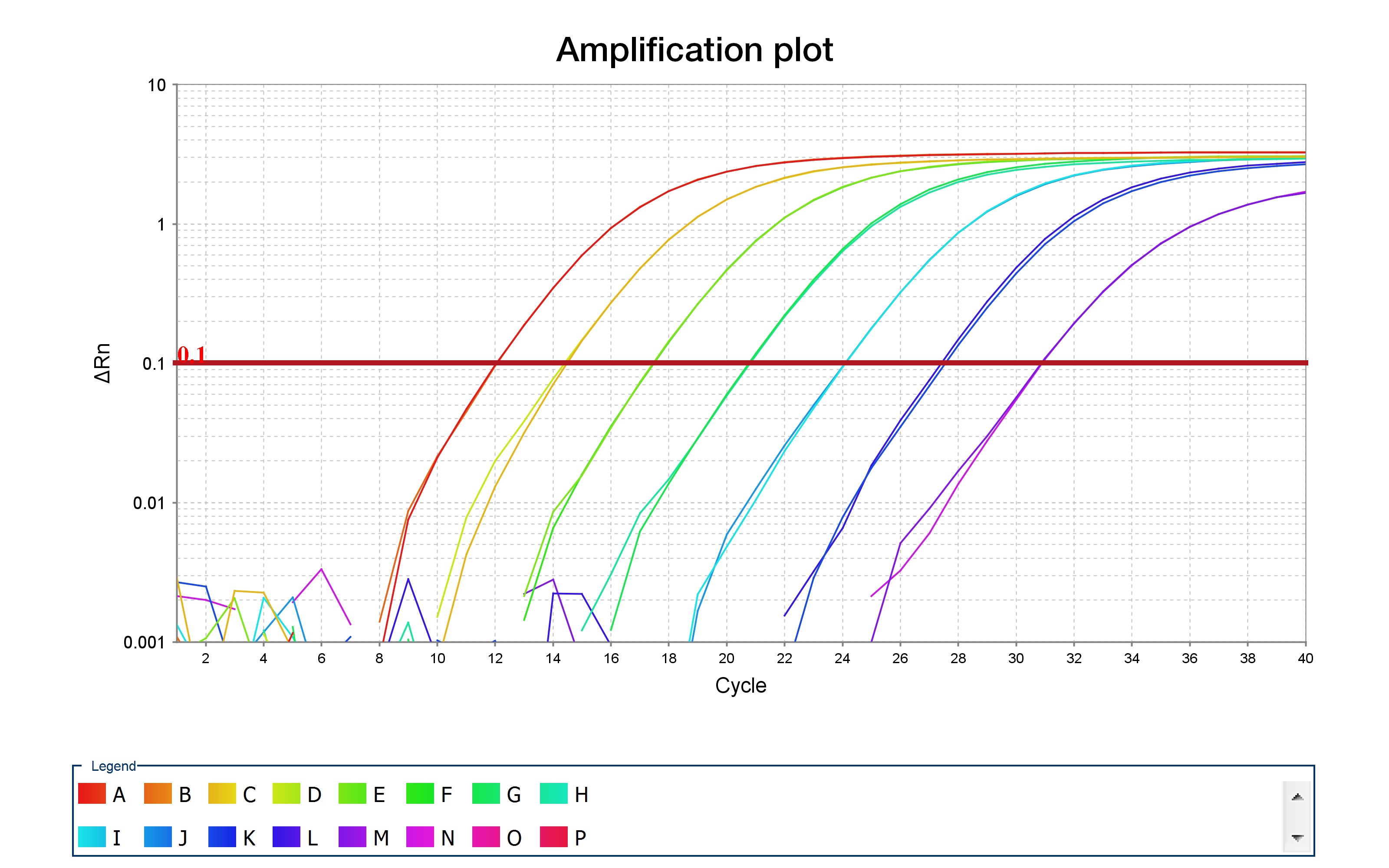
After some number of cycles (given on the x-axis), the amplification profile begins to increase until it reaches saturation. The position on the x-axis should be displaced between your samples -- "blank" samples should not amplify at all, while samples with 1ng of DNA should have an amplification curve which is delayed when compared to samples containing 10ng of DNA.
By looking at these amplification curves, it is straightforward to determine the "optimal" number of PCR cycles to use for barcoding. Choose the number of cycles that correspond to the mid-point of the curves, for each primer pair, with 10ng of DNA. As an example, consider the red curve in the amplification plot above. The midpoint of this curve corresponds to approximately 10 cycles. Thus, you should use 10 cycles when performing your barcoding PCR. This is to ensure that DNA levels do not saturate, which would bias your PCR amplifications when barcoding.
After performing qPCR, the next step is to perform the barcoding PCRs. This is done by setting up PCR reactions in precisely the same way, with the same primers, but with 2x Q5 polymerase mix rather than the 2x qPCR mix. Depending on your amplification results from the qPCR experiment, you should decide whether you'd like to use 1ng template or 10ng template; just be consistent and use the same DNA concentration across all primer sets. Program the thermocycler with the following settings:
| cycles | temperature | time |
|---|---|---|
| 1 | 98°C | 30 seconds |
| see qPCR results | 98°C | 10 seconds |
| see qPCR results | (anneal) | 30 seconds |
| see qPCR results | 72°C | 30 seconds |
| 1 | 72°C | 120 seconds |
| Hold | 4°C | ∞ |
After amplifying the oligo libraries for EACH PRIMER PAIR, perform a gel extraction by adding 10 microliters of 6x NEB DNA dye to each 50 microliter PCR reaction. Load the full volumes on a thick, 2% agarose gel. Perform electrophoresis for 45 minutes at 120V. Use a scalpel to remove the DNA band corresponding to the amplified oligo libraries. Perform a gel extraction using one of many commercially-available kits. We have previously obtained good results with the Zymoclean Gel DNA Recovery Kit.
Amplification of oligo pools must account for several considerations. First, the reverse primer in the barcoding PCR should add the 20nt, random barcode sequence. Such a primer can be ordered from IDT by inputting 'NNNNNNNNNNNNNNNNNNNN' in the desired barcode position. The reverse primer must also have a region that overlaps the template; shoot for an annealing temperature between 61°C - 64°C. The reverse primer must also include a sequence to be used for cloning into the desired plasmid later (via Golden Gate or Gibson assembly).
Rev should have: primer sequence that binds to the oligo pool (specific to each primer set), the barcode, a fixed sequence that can be used for downstream sequencing, and a sequence that is used for cloning into the desired plasmid, either Gibson or Golden Gate).
Fwd should have: primer sequence that binds to the oligo pool (specific to each primer set), a fixed sequence that can be used for downstream sequencing, and a sequence that is used for cloning into the desired plasmid, either Gibson or Golden Gate).
Discuss both Gibson and Golden Gate options, XhoI, SbfI, SalI, etc. and need for dephosphorylation and everything else. See NEB site. Post Benchling links to plasmid, and also discuss genome-integration option.
The barcode was inserted 110 base pairs from the 5’ end of the mRNA, containing 45 base pairs from the targeted regulatory region, 64 base pairs containing primer sites used in the construction of the plasmid, and 11 base pairs containing a three frame stop codon. All the sequences are listed in Supplementary Table 1. Following the barcode there is an RBS and a GFP coding region.
[FIND SUPPLEMENTARY TABLE 1 FROM THE PAPER, GO THRU THOSE SEQUENCES TO HELP EXPLAIN THIS STUFF]
After amplifying, barcoding, and purifying the variety of oligo pools, the next step is to insert each of these library "groups" into a plasmid, which can then be cloned into E. coli. In prior Reg-Seq experiments, all oligo pools were cloned and expressed from a plasmid; they were not genome-integrated. For the purposes of this protocol, we will discuss both plasmid expression and a potential method for genome-integration.
Reg-Seq experiments have been performed by cloning oligo libraries into pJK14 plasmid (SC101 origin) via Gibson assembly. Overhang "arms" from the PCR amplicons with homology to this plasmid were used to insert the oligo pools. To use Gibson assembly for cloning PCR-amplified, barcoded oligo pools into pJK14, you should first amplify the backbone using the primer-binding sites specified on the Benchling sequence (https://benchling.com/s/seq-M9lQusDbSzsjmGihPxYr). See the pink annotations on the Benchling sequence for the Gibson sites and primer amplification binding sites. This plasmid encodes kanamycin resistance.
Perform Gibson assembly according to NEB's manufacturer instructions. Prior to electroporation, perform drop dialysis with water for at least 30 minutes. Electroporate into highly electrocompetent DH5α cells (these can be purchased from NEB), shooting for a time constant of electroporation (1800 mV) exceeding 5.0 milliseconds. If desired, one can also electroporate directly into the strain to be studied (E. coli K-12 MG1655), but it is typically a good idea to first electroporate into a highly competent strain, isolate the library again, perform routine checks (e.g. gel electrophoresis and so forth), and then transform into the final strain. This also enables one to store the isolated, cloned DNA library for future use.
In future iterations of Reg-Seq, it might be a good idea to clone promoter mutant libraries directly into the genome of E. coli. Lambda red recombination is not an efficient process, however, and thus would highly bias the library of interest. Therefore, we have previously tested other methods, especially those used by the Kosuri lab at UCLA.
In our lab, we have a plasmid called pLibAcceptorV2 (Addgene link) which can be used to genome-integrate any DNA sequences cloned onto this plasmid into a genetic locus of interest. This method requires two important steps, however:
- The accepting strain of E. coli (K-12 MG1655) must first be genomically modified, using classical recombination, by inserting a "landing pad" at the genomic position of interest. Specifically, lox sites must be inserted into the genome position of interest, which correspond to lox sites on the pLibAcceptorV2 plasmid. lox71 recombines with lox66. Note that the genomic landing pad sequences are reversed, so one must consider their positioning if concerned with the orientation of the reporter after integration.
Construct Site Sequences (Spacer in bold)
| Construct | Site | Sequence (spacer in bold) |
|---|---|---|
| pLibAcceptorV2 | lox66 | 5'- ATAACTTCGTATAGCATACATTATACGAAcggta -3' |
| pLibAcceptorV2 | _lox_m2/71 | 5'- taccgTTCGTATATGGTTTCTTATACGAAGTTAT -3' |
| Genomic Landing Pad | _lox_m2/66 (reverse orientation) | 5'- taccgTTCGTATAAGAAACCATATACGAAGTTAT -3' |
| Genomic Landing Pad | _lox_71 (reverse orientation) | 5'- ATAACTTCGTATAATGTATGCTATACGAAcggta -3' |
The landing pad sequence can be found at this Benchling link: https://benchling.com/s/seq-Kl6BOcb78yxDkgxwi2ha // It just contains constitutively active mCherry & Chloramphenicol resistance flanked by complementary loxP sites. Insert this into the genomic position of interest.
The pLibAcceptorV2 plasmid (https://benchling.com/s/seq-8ros6uaUuTsZIwPMir8m) contains an arabinose-inducible Cre-recombinase, a heat curable ori (to remove plasmid after cre-lox cassette exchange), and a selectable marker/library cloning site flanked by loxP sites. Built around the library cloning site are priming sites for sequencing as well as terminators to block outside transcriptional interference. The plasmid also has restriction sites built-in for restriction-based cloning of oligo libraries.
If you wish to perform genome-integration for oligo library cloning, you should first purify ample concentration of pLibAcceptorV2 plasmid by growing cells expressing this plasmid at 30°C for 24 hours, and then performing a maxiprep to isolate plasmid (which is very low-copy). After maxiprep, clean the plasmid further by using a Zymo DNA Clean & Concentrator-25 kit. This ensures that genomic DNA, which can sometimes "bleed" through a maxiprep, is fully removed and solely plasmid remains.
Cell libraries were then grown to saturation in LB and diluted 1:10,000 into the appropriate growth media for the promoter under consideration, and grown to an optical density at 600 nm of 0.2–0.4. A Beckman Coulter MoFlo XDP cell sorter was used to sort cells by fluorescence, with 500,000 cells collected into each of the four bins. Sorted cells were then regrown overnight in 10 mL of LB media under kanamycin selection. The plasmids in each bin were miniprepped (Qiagen) after overnight growth, and PCR was used to amplify the mutated region from each plasmid for Illumina sequencing. SI Appendix has additional details on library construction and Sort-Seq as well as on calculating expression shift plots and energy matrices.
Dilution also (see Guillaume protocols).
Cells were grown to an optical density of 0.3 and RNA was then stabilized using Qiagen RNA Protect (Qiagen, Hilden, Germany). Lysis was performed using lysozyme (Sigma Aldrich, Saint Louis, MO) and RNA was isolated using the Qiagen RNA Mini Kit. Reverse transcription was performed using Superscript IV (Invitrogen, Carlsbad, CA) and a specific primer for the labeled mRNA.
The growth conditions studied in this study were inspired by [1] and include differing carbon sources such as growth in M9 with 0.5% Glucose, M9 with acetate (0.5%), M9 with arabinose (0.5%), M9 with Xylose (0.5%) and arabinose (0.5%), M9 with succinate (0.5%), M9 with fumarate (0.5%), M9 with Trehalose (0.5%), and LB. In each case cell harvesting was done at an OD of 0.3. These growth conditions were chosen so as to span a wide range of growth rates, as well as to illuminate any carbon source specific regulators. We also used several stress conditions such as heat shock, where cells were grown in M9 and were subjected to a heat shock of 42 degrees for 5 minutes before harvesting RNA. We grew in low oxygen conditions. Cells were grown in LB in a container with minimal oxygen, although some will be present as no anaerobic chamber was used. This level of oxygen stress was still sufficient to activate FNR binding, and so activated the anaerobic metabolism. We also grew cells in M9 with Glucose and 5mM sodium salycilate. Growth with zinc was preformed at a concentration of 5mM ZnCl2 and growth with iron was performed by first growing cells to an OD of 0.3 and then adding FeCL2 to a concentration of 5mM and harvesting RNA after 10 minutes. Growth without cAMP was accomplished by the use of the JK10 strain which does not maintain its cAMP levels.
When are 4nt barcodes for each growth condition added? This must be after the mapping phase...drop in GFP with variable, 4nt barcodes?
Details here.