Ring Simulation
In this section we'll see how to make the GDIS simulation files included with Diffraction Ring Profiler.
GDIS(Graphical Display Interface for Structures) can simulate electron powder diffraction from CIF files.
GDIS Home Page: http://gdis.sourceforge.net/
CIF files for crystals are commonly available and can be created with Crystal Maker or Vesta. In this section we'll use Vesta which is available for download from the JP-minerals website.
Vesta Home Page: http://jp-minerals.org/vesta/en/
If you already have a CIF file for your structure then you can skip to the section about GDIS.
To make the crystal structure you need to know the space group and lattice parameters of your structure. Click File -> New structure and go to the Unit cell tab then enter the space group and lattice parameters of your structure. Gold is cubic Fm-3m with a 4.0782 Å lattice parameter:
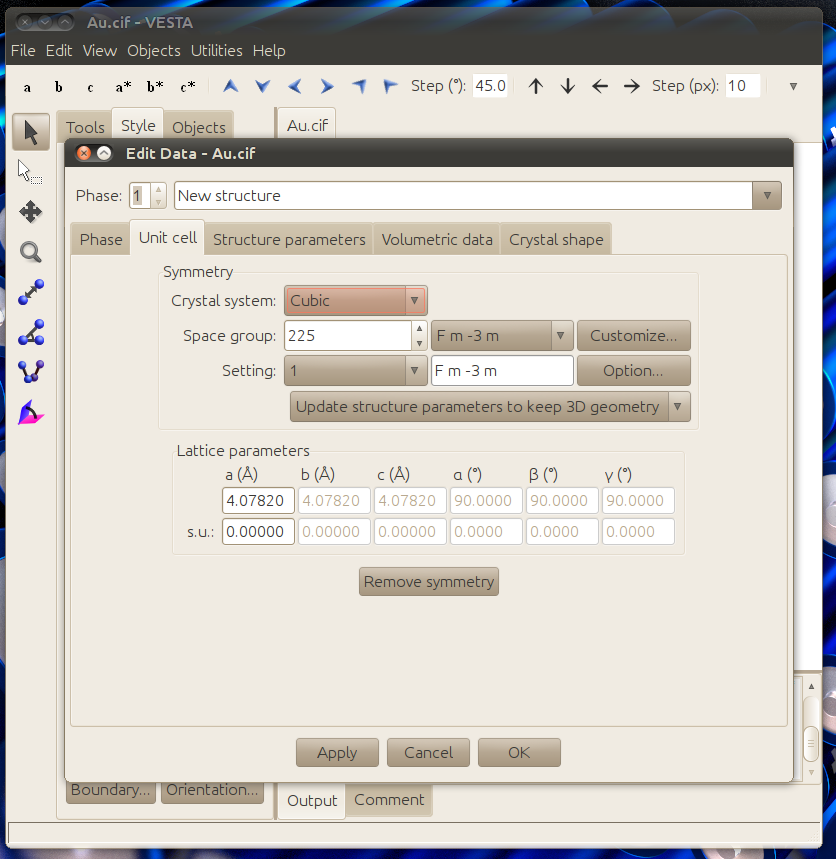
In the structure parameter tab make a new atom of Au and place it in the 0,0,0 symmetry location.

The resulting structure will look like this:
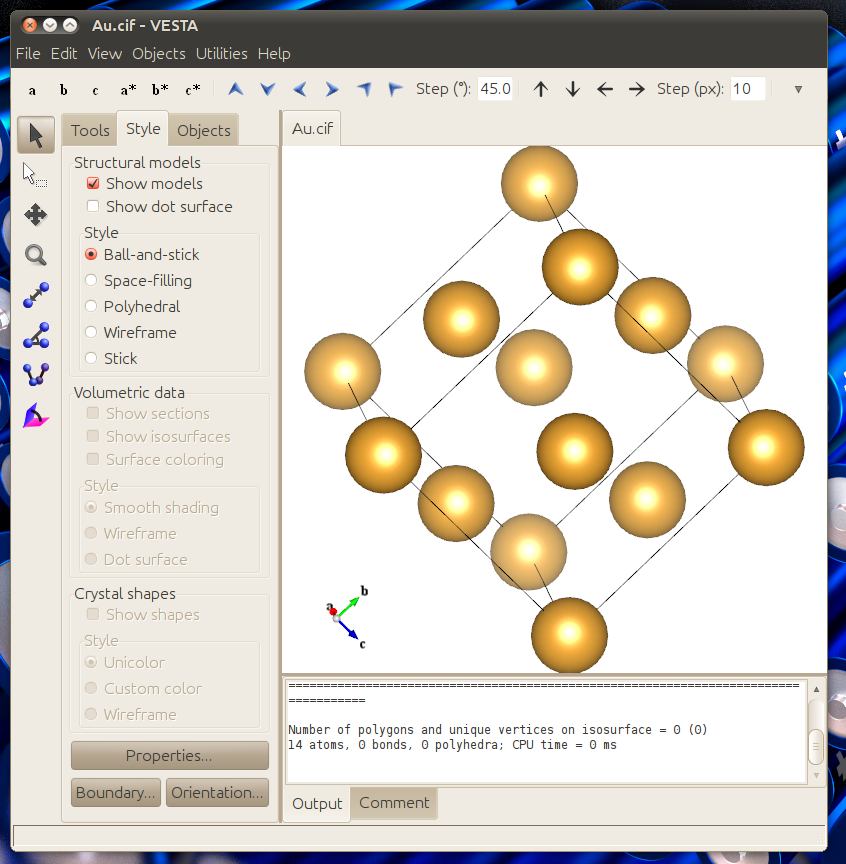
Then click File -> Export Data... and then save the structure as a .cif file.
Open your Au CIF file that was made in the previous step or with another software package with GDIS by clicking File -> Open.. and selecting the file. It will open to look like this.
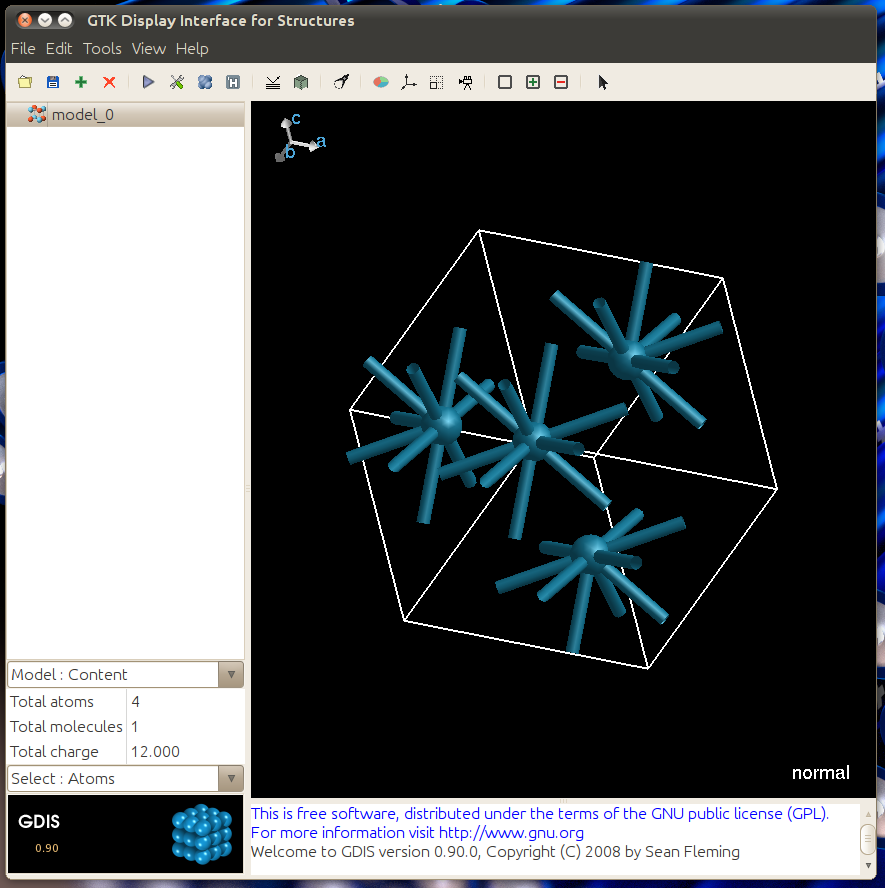
Then click on the this bottom to simulate a diffraction.
Match the dialogue to the one below to get a peak simulation.
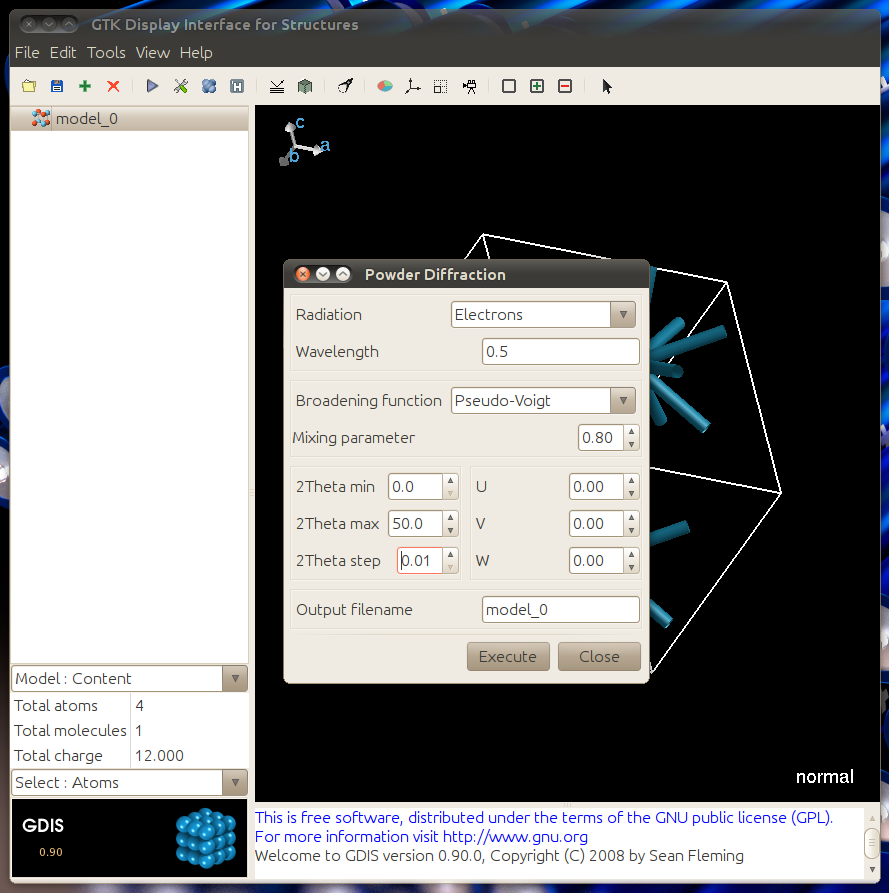
The resulting graph should match the one below. The graph data can be exported by using File -> Export -> Graph Data. Save the file with a .txt extension.

Match the dialogue to the one below to get a profile simulation. You can adjust the parameters to match the broadening typically seen in your sample.
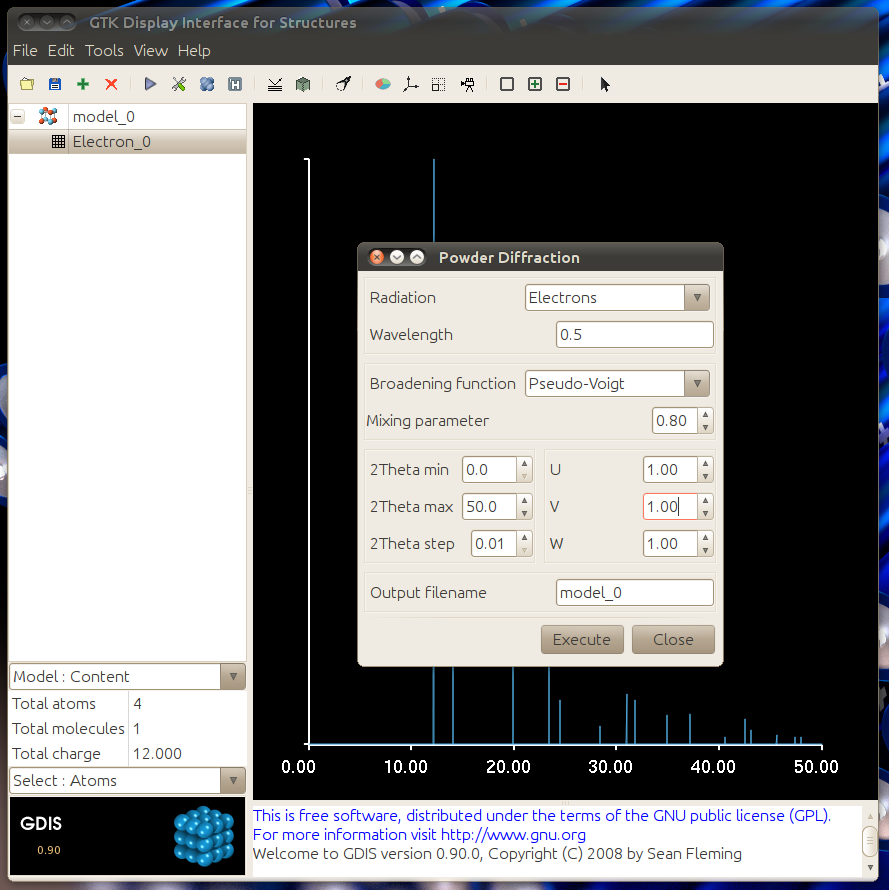
The resulting graph should match the one below. The graph data can be exported by using File -> Export -> Graph Data. Save the file with a .txt extension.
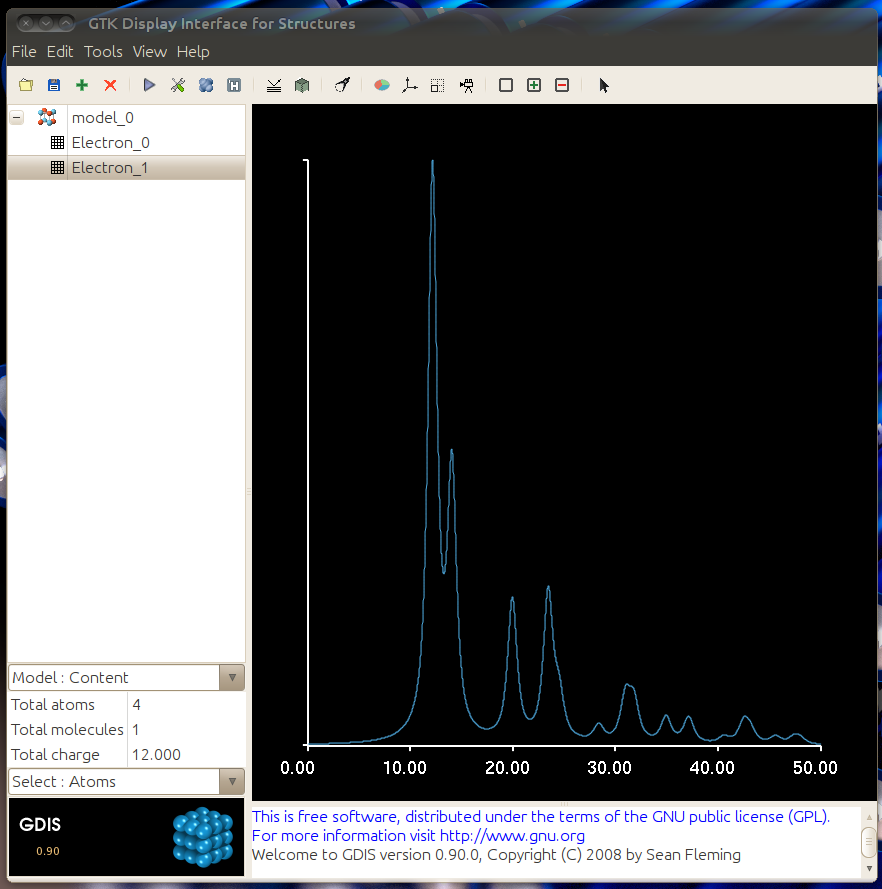
These two .txt files can be used with Diffraction Ring Profiler to match diffraction ring pattern peak positions and profiles to the simulated ideal crystal structure.