GCP_RAPT_doc
As of December 2024, NCBI's pilot tool, Read assembly and Annotation Pipeline (RAPT) tool will no longer be available. We encourage you to check out NCBI’s suite of assembly and annotation tools including the genome assembler SKESA, the taxonomic assignment tool ANI, and the prokaryotic genome annotation pipeline (PGAP). Learn more...
This page contains the prerequisites and instructions for running GCP RAPT, RAPT for the Google Cloud Platform, using the run_rapt_gcp.sh script.
Some basic knowledge of Unix/Linux commands, SKESA, and PGAP is useful.
Please see our wiki page for References, Licenses and FAQs.
-
System Requirements
-
Get the RAPT command-line interface
- Try RAPT
-
Review the output
-
Additional information
-
run_rapt_gcp.sh usage examples
GCP RAPT is designed to run on the Google Cloud Platform (GCP). The script run_rapt_gcp.sh takes care of launching the instance on which RAPT will run, and shutting it down after RAPT finishes. Therefore, it can conveniently be invoked from the Google Cloud Shell or from any Google instance meeting the following minimal requirements:
- gcloud SDK installed (automatically enabled in Cloud Shell)
- gsutil tool installed (automatically enabled in Cloud Shell)
- Cloud Life Sciences API enabled for your project. To do this:
-
In a browser, sign into GCP with your Google username and password, and select the project where RAPT will run.
-
Enable the Cloud Life Sciences API for the project. This only needs to be done once, using the following steps:
-
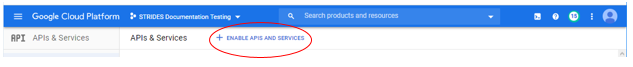
Type "api" in the "Search products and resources" box and select "APIs & Services"

-
Select ENABLE APIS AND SERVICES
-
Type "cloud life" in the search box and select "Google Cloud Life Sciences API"

-
Choose Enable, use all defaults. You are now finished with this operation.
-
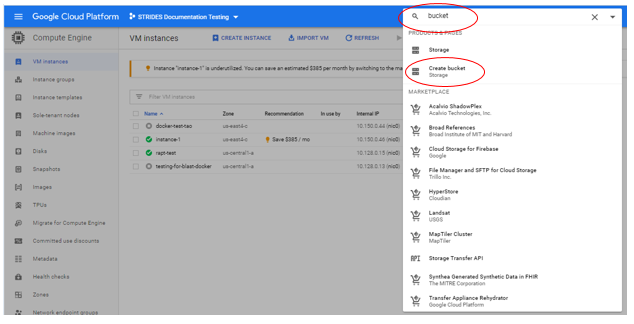
- Access to a GCP storage bucket for the data RAPT will produce. To create a bucket:
-
Type "bucket" in the "Search products and resources" box and select "Create bucket"
-
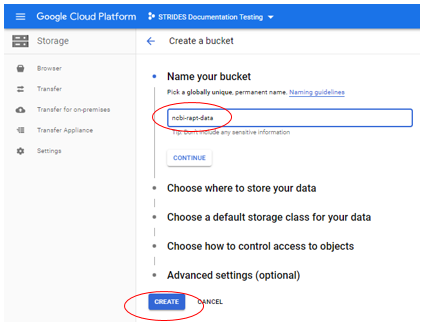
Name your bucket and select CREATE
Note: By choosing Create, you bucket is personal, if you want to share it with others, you can make modifications to its permissions after it is created.
-
- In a browser, sign into GCP with your google username and password, and select the project where RAPT will run.
- Invoke a Cloud Shell
- At your Cloud Shell prompt, download the latest release by executing the following commands:
~$ curl -sSLo rapt.tar.gz https://github.com/ncbi/rapt/releases/download/v0.5.5/rapt-v0.5.5.tar.gz
~$ tar -xzf rapt.tar.gz && rm -f rapt.tar.gz
Now you should have the run_rapt_gcp.sh script in your current directory.
Running run_rapt_gcp.sh with the help subcommand will provide instructions and options to run GCP RAPT.
~$ ./run_rapt_gcp.sh help
Usage: run_rapt_gcp.sh <command> [options]
Job creation commands:
submitacc <sra_acxn> <-b|--bucket URL> [--label LABEL]
[--skesa-only] [--no-usage-reporting] [--stop-on-errors] [--machine-type TYPE]
[--boot-disk-size NUM] [--timeout SECONDS]
Submit a job to run RAPT on an SRA run accession (sra_acxn).
submitfastq <fastq_uri> <--organism "Genus species"> [--strain "ATCC xxxx"]
<-b|--bucket URL> [--label LABEL] [--skesa-only]
[--no-usage-reporting] [--stop-on-errors] [--machine-type TYPE] [--boot-disk-size NUM]
[--timeout SECONDS]
Submit a job to run RAPT on Illumina reads in FASTQ or FASTA format.
fastq_uri is expected to point to a google cloud storage (bucket). If forward
and reverse readings are in two separate files, put them in the same storage
bucket and delimit their names with a comma (no space!) in fastq_uri:
submitfastq gs://mybucket/forward.fastq,reverse.fastq
The --organism argument is mandatory. It is the binomial name or, if the
species is unknown, the genus for the sequenced organism. This identifier
must be valid in NCBI Taxonomy. The --strain argument is optional.
test <-b|--bucket> [--label LABEL] [--skesa-only]
[--no-usage-reporting] [--stop-on-errors]
Run a test suite. When RAPT does not produce the expected results, it may be
helpful to use this command to ensure RAPT is functioning normally.
Common options:
======================
-b|--bucket URL
Mandatory. Specify the destination storage location to store results
and job logs.
--label LABEL
Optional. Tag the job with a custom label, which can be used to filter jobs
with the joblist command. Google cloud platform requires that the label can
only contain lower case letters, numbers and dash (-). Dot and white spaces
are not allowed.
--skesa-only
Optional. Only assemble sequences to contigs, but do not annotate.
--no-usage-reporting
Optional. Prevents usage report back to NCBI. By default, RAPT sends usage
information back to NCBI for statistical analysis. The information collected
are a unique identifier for the RAPT process, the machine IP address, the
start and end time of RAPT, and its three modules: SKESA, taxcheck and PGAP.
No personal or project-specific information (such as the input data) are
collected.
--stop-on-errors
Optional. Do not run PGAP annotation pipeline when the genome sequence is
misassigned or contaminated
--auto-correct-tax
Optional. If the genome sequence is misassigned or contaminated and ANI predicts
an organism with HIGH confidence, use it for PGAP instead of the one provided by
the user.
--regions
Optional, comma-separated. Specify in which GCP region(s) RAPT should run.
Note: it should be regions in which you have sufficient CPU quotas (verify
at https://console.cloud.google.com/iam-admin/quotas/details). Default is
a single region, us-east4.
--machine-type TYPE
Optional. Specify the type of google cloud virtual machine to run this job
(see Google documentation, https://cloud.google.com/compute/docs/machine-types).
Default is "n1-highmem-8", which is suitable for most jobs.
--boot-disk-size NUM
Optional. Set the size (in Gb) of boot disk for the virtual machine. Default
size is 128.
--timeout SECONDS
Optional. Set the timeout (seconds) for the job. Default is 86400s
(24 hours).
Job control commands:
joblist [-n|--limit NUM] [--csv]
[-d|--delimiter DELIM]
List jobs under current project. Jobs are are sorted by their submission time,
latest first. Use -n or --limit to limit the number of jobs to display.
Specify --csv will list jobs in comma-delimited table instead of
tab-delimited. Specify a delimit character using "-d|--delimiter DELIM"
will override --csv and output a custom delimited table.
jobdetails <job-id>
All job creating commands, if successful, will display a job-id that uniquely
identify the job created. This command can be used to display the detailed
information of the job identified by the job-id. Be aware this is mostly about
technical details of how the job is created and handled by google cloud platform,
mostly useful to developers and technical staff than to general users.
cancel <job-id>
Cancel a running job
version
Display the current RAPT version.
help
Display this usage page.To run RAPT, you need the Illumina-sequenced reads for the genome you wish to assemble and annotate. These can be in a fasta or fastq file available locally or in a Google bucket, or they can be in the NCBI Sequence Read Archive (SRA).
Important:
- Only reads sequenced on Illumina machines can be used by RAPT.
- The reads provided should be from a single isolate.
To demonstrate how to run RAPT, we are going to use SRR3496277, a set of reads available in SRA for Mycoplasma pirum. This example takes about 20 minutes to complete.
Run the following command, where gs://your_results_bucket is a GCP bucket for which you have write permission.
~$ ./run_rapt_gcp.sh submitacc SRR3496277 --bucket gs://your_results_bucketThe script will print out execution information similar to the following:
```bash
~$ ./run_rapt_gcp.sh submitacc SRR3496277 --bucket gs://your_results_bucket
RAPT job has been created successfully.
----------------------------------------------------
Job-id: 5541b09bb9
Output storage: gs://your_results_bucket/5541b09bb9
GCP account: 1111111111111-compute@developer.gserviceaccount.com
GCP project: example
----------------------------------------------------
[**Attention**] RAPT jobs may take hours to finish. Progress of this job can be viewed in GCP stackdriver log viewer at:
https://console.cloud.google.com/logs/viewer?project=strides-documentation-testing&filters=text:5541b09bb9
For current status of this job, run:
run_rapt_gcp.sh joblist | fgrep 5541b09bb9
For technical details of this job, run:
run_rapt_gcp.sh jobdetails 5541b09bb9
~$
```
The results for the job will be available in the bucket you specified after the job is marked 'Done'. Please note that some jobs may take up to 12 hours.
To run RAPT with file inputs, use the submitfastq command to provide the location of the input file(s), and specify the the organism name and the Google bucket where the output should be placed.
- Data should be short reads, from an Illumina sequencer.
- Both single- or paired-end reads are acceptable.
- Paired-end reads can be provided in one file with the two reads of a pair adjacent to each other (interleaved) in the file, or as two files, with the forward reads in one file and the reverse reads in the other.
- The files should be in fastq or fasta format.
- The quality scores are not necessary.
- The files may be gzipped.
- The files need to be available in a Google storage bucket, or locally to the Google Cloud shell, or on the instance on which you run
run_rapt_gcp.sh.
There are mutiple ways of doing this. Here are two:
-
Upload a file to a GCP bucket
Select upload from GCP bucket screen

-
Upload function from your Cloud Shell.

The organism name assigned to the reads needs to be provided with the --organism parameter. It can be the genus species, or the genus only if the species is not defined. The organism name must be known to the NCBI Taxonomy. The strain can optionally be provided with the --strain parameter. If you are unsure about the name to provide, use the flag auto-correct-tax or stop-on-errors (see more details in Useful options).
Here is an example command using the file myreads.fastq located in the bucket my_input_bucket. The results will be copied to the bucket my_results_bucket when the job finishes:
~$ ./run_rapt_gcp.sh submitfastq gs://my_input_bucket/myreads.fastq -b gs://my_results_bucket --label M_pirum_25960 --organism "Mycoplasma pirum" --strain "ATCC 25960"Or, using separate files of forward and reverse reads located in the bucket my_input_bucket:
~$ ./run_rapt_gcp.sh submitfastq gs://my_input_bucket/myforwardreads.fastq,myreversereads.fastq -b gs://my_results_bucket --label M_pirum_25960 --organism "Mycoplasma pirum" --strain "ATCC 25960"Or, using local gzipped fasta files of forward and reverse reads (no space after comma):
~$ ./run_rapt_gcp.sh submitfastq ./myforwardreads.fasta.gz,./myreversereads.fasta.gz -b gs://my_results_bucket --label M_pirum_25960 --organism "Mycoplasma pirum" --strain "ATCC 25960"Once the job is launched, the script will print out execution information similar to the following:
RAPT job has been created successfully.
----------------------------------------------------
Job-id: b2ac02d7c7
Output storage: gs://your_results_bucket/b2ac02d7c7
GCP account: 1111111111111-compute@developer.gserviceaccount.com
GCP project: example
----------------------------------------------------
[**Attention**] RAPT jobs may take hours to finish. Progress of this job can be viewed in GCP stackdriver log viewer at:
https://console.cloud.google.com/logs/viewer?project=strides-documentation-testing&filters=text:b2ac02d7c7
For current status of this job, run:
run_rapt_gcp.sh joblist | fgrep b2ac02d7c7
For technical details of this job, run:
run_rapt_gcp.sh jobdetails b2ac02d7c7
~$
The results for the job will be available in the bucket you specified after the job is marked 'Done'. Please note that the duration of the job is highly dependent on the size of the genome and may be of several hours.
-
--auto-correct-tax:
When set: if the step that evaluates the taxonomic assignment of the assembly (see the Average Nucleotide Identity tool) determines with high confidence that the species that best fits your assembly is different from the name you or SRA provided on input, the annotation process will use the ANI-chosen scientific name, and the ANI-chosen name will appear in the output files. When not set (default): the annotation process will use the name provided on input, regardless of the results of ANI. -
--stop-on-errors:
When set: if the step that evaluates the taxonomic assignment of the assembly (see the Average Nucleotide Identity tool) determines with high confidence that the species that best fits your assembly is different from the name you provided or SRA on input, or that the assembly is contaminated, RAPT will stop after the taxonomy check and no annotation results will be produced.
GCP RAPT generates a tarball named output.tar.gz in your designated bucket, under a "directory" named after the 10-character job-id assigned at the start of the execution (i.e. "b2ac02d7c7"). The tarball contains the following files:
-
Assembly results:
- skesa_out.fa: multifasta files of the assembled contigs produced by SKESA
- assembly_stat_report.tsv: assembly statistics (contig count, base count, min contig length, max contig length, contig N50, contig L50)
- skesa_out.fa: multifasta files of the assembled contigs produced by SKESA
-
Taxonomy verification (see more details):
- ani-tax-report.txt: results of the taxonomy check on the assembled sequences reported by the Average Nucleotide Identity tool, in text format. The possible statuses are:
CONFIRMED: The organism name associated with the reads, and assigned to the genome, has been confirmed by ANI. The submitted genus matches the genus of the ANI-predicted organism.
MISASSIGNED: The organism has been found to be misassigned to the genome. The submitted genus does not match the genus of the ANI-predicted organism.
INCONCLUSIVE: The organism cannot be identified (due to lack of a close enough type assembly in GenBank)
CONTAMINATED: The genome is contaminated with sequences from an organism other than the submitted organism.
- ani-tax-report.xml: same as above, in XML format
- ani-tax-report.txt: results of the taxonomy check on the assembled sequences reported by the Average Nucleotide Identity tool, in text format. The possible statuses are:
-
PGAP annotation results in multiple formats (see a detailed description of the annotation output files):
- annot.gbk: annotated genome in GenBank flat file format
- annot.gff: annotated genome in GFF3 format
- annot.sqn: annotated genome in ASN format
- annot.faa: multifasta file of the proteins annotated on the genome
- calls.tab: tab-delimited file of the coordinates of detected foreign sequence. Empty if no foreign contaminant was found.
- annot.gbk: annotated genome in GenBank flat file format
-
CheckM completeness results:
- checkm.txt: Annotated assembly completeness and contamination as calculated by CheckM. See a full description of the file format at this location. Note: 1) The CheckM calculation is performed on the proteins produced by PGAP, 2) the set of markers used by CheckM is determined by the species associated with the genome (as provided on input or as overridden by ANI).
- checkm.txt: Annotated assembly completeness and contamination as calculated by CheckM. See a full description of the file format at this location. Note: 1) The CheckM calculation is performed on the proteins produced by PGAP, 2) the set of markers used by CheckM is determined by the species associated with the genome (as provided on input or as overridden by ANI).
-
Execution logs:
- concise.log: log file containing the major events in a run
- verbose.log: all messages, with time stamps
- concise.log: log file containing the major events in a run
Along with the tarball, a run.log file generated automatically by the Google Life Sciences Pipeline will also be in the bucket. This file catches all output to stdout and stderr by anything, and may be helpful to identify the problem should any happens.
- RAPT will be unable to submit jobs if GCloud configuration provides a default region. Please create a new Google Cloud configuration without specifying a default region to run this script.
- Google Storage Bucket URI - Required for each run. The user needs to provide a Google Storage Bucket uri for storing the logging and output file. One can provide the bucket uri by specifying --bucket argument when submit the job, or one can provide the bucket uri by setting the BUCKET environment variable.
or:
~$ ./run_rapt_gcp.sh --bucket gs://your-bucket/path ...~$ export BUCKET="gs://your-bucket/path" ~$ ./run_rapt_gcp.sh command ...
- The joblist command shows a list of jobs with headers as follow: JOB_ID, USER, LABEL, SRR, STATUS, START_TIME, END_TIME, OUTPUT_URI. STATUS Done represents a completed job.
- A Cloud Shell has certain limits, if you find these limits too restricting you can create a Virtual Machine. When creating the Virtual Machine, make sure you select the option to "Allow full access to all Cloud APIs".
- Check the details of a job:
~$ ./run_rapt_gcp.sh jobdetails job-id
- List all jobs for the project in table format:
~$ ./run_rapt_gcp.sh joblist - List the latest N jobs in tabular format:
~$ ./run_rapt_gcp.sh joblist -n N - List the latest N jobs in tabular format:
~$ ./run_rapt_gcp.sh joblist -n N --csv
- Cancel a running job:
./run_rapt_gcp.sh cancel job-id
- Run test cases without waiting the for the test to complete:
~$ ./run_rapt_gcp.sh test --bucket gs://your-bucket
If you have other questions, please visit our [FAQs page](FAQ).