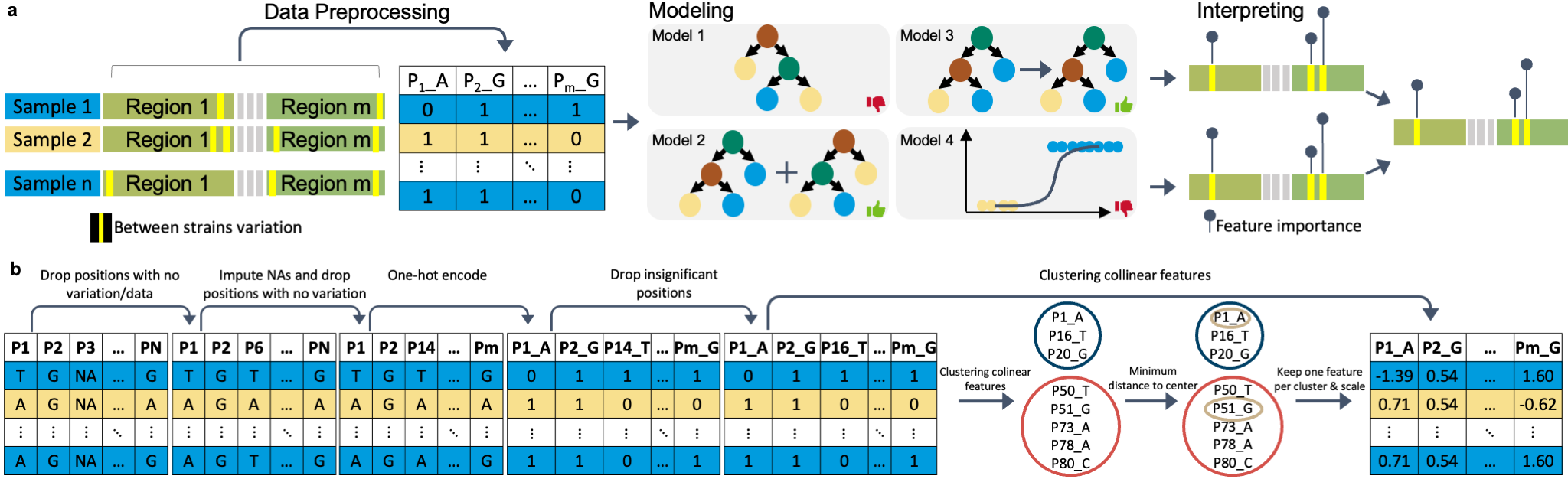
deepBreaks , a computational method, aims to identify important changes in association with the phenotype of interest using multi-alignment sequencing data from a population.
Key features:
- Generality: deepBreaks is a new computational tool for identifying genomic regions and genetic variants significantly associated with phenotypes of interest.
- Validation: A comprehensive evaluation of deepBreaks performance using synthetic data generation with known ground truth for genotype-phenotype association testing.
- Interpretation: Rather than checking all possible mutations (breaks), deepBreaks prioritizes only statistically promising candidate mutations.
- Elegance: User-friendly, open-source software allowing for high-quality visualization and statistical tests.
- Optimization: Since sequence data are often very high volume (next-generation DNA sequencing reads typically in the millions), all modules have been written and benchmarked for computing time.
- Documentation: Open-source GitHub repository of code complete with tutorials and a wide range of real-world applications.
Citation:
Mahdi Baghbanzadeh, Tyson Dawson, Bahar Sayoldin, Seth A. Frazer, Todd H. Oakley, Keith A. Crandall, Ali Rahnavard (2023). deepBreaks: a machine learning tool for identifying and prioritizing genotype-phenotype associations , https://github.com/omicsEye/deepBreaks/.
- Generic software that can handle any kind of sequencing data and phenotypes
- One place to do all analysis and producing high-quality visualizations
- Optimized computation
- User-friendly software
- Provides a predictive power of most discriminative positions in a sequencing data
- First install conda
Go to the Anaconda website and download the latest version for your operating system. - For Windows users: do not forget to add
condato your systempath - Second is to check for conda availability
open a terminal (or command line for Windows users) and run:
conda --version
it should out put something like:
conda 4.9.2
if not, you must make conda available to your system for further steps. if you have problems adding conda to PATH, you can find instructions here.
If you are using an Apple M1/M2 MAC please go to the Apple M1/M2 MAC for installation
instructions.
If you have a working conda on your system, you can safely skip to step three.
If you are using windows, please make sure you have both git and Microsoft Visual C++ 14.0 or greater installed.
install git
Microsoft C++ build tools
In case you face issues with this step, this link may help you.
- Create a new conda environment (let's call it deepBreaks_env) with the following command:
conda create --name deepBreaks_env python=3.9
- Activate your conda environment:
conda activate deepBreaks_env
- Install deepBreaks: install with pip:
pip install deepBreaks
or you can directly install if from GitHub:
python -m pip install git+https://github.com/omicsEye/deepbreaks
- Update/install Xcode Command Line Tools
xcode-select --install
- Install Brew
/bin/bash -c "$(curl -fsSL https://raw.githubusercontent.com/Homebrew/install/HEAD/install.sh)"
- Install libraries for brew
brew install cmake libomp
- Install miniforge
brew install miniforge
- Close the current terminal and open a new terminal
- Create a new conda environment (let's call it deepBreaks_env) with the following command:
conda create --name deepBreaks_env python=3.9
- Activate the conda environment
conda activate deepBreaks_env
- Install packages from Conda
conda install -c conda-forge lightgbm
pip install xgboost
- Finally, install deepBreaks: install with pip:
pip install deepBreaks
or you can directly install if from GitHub:
python -m pip install git+https://github.com/omicsEye/deepbreaks
If you are using Docker, you can pull the image from Docker Hub:
docker pull omicseye/deepbreaks-dc:latest
For instructions on how to use the Docker image, please visit the Docker Hub page.
To test if deepBreaks is installed correctly, you may run the following command in the terminal:
deepBreaks -h
Which yields deepBreaks command line options.
$ deepBreaks -h
usage: deepbreaks [-h] --seqfile SEQFILE --seqtype SEQTYPE --meta_data
META_DATA --metavar METAVAR [--gap GAP]
[--miss_gap MISS_GAP] [--ult_rare ULT_RARE] --anatype
{reg,cl}
[--distance_metric {correlation,hamming,jaccard,normalized_mutual_info_score,adjusted_mutual_info_score,adjusted_rand_score}]
[--fraction FRACTION]
[--redundant_threshold REDUNDANT_THRESHOLD]
[--distance_threshold DISTANCE_THRESHOLD]
[--top_models TOP_MODELS] [--aggregate AGGREGATE] [--cv CV]
[--test TEST] [--ref_id REF_ID] [--ref_compare REF_COMPARE]
[--compare_len COMPARE_LEN] [--tune] [--plot] [--write]
optional arguments:
-h, --help show this help message and exit
--seqfile SEQFILE, -sf SEQFILE
files contains the sequences
--seqtype SEQTYPE, -st SEQTYPE
type of sequence: 'nu' for nucleotides or 'aa' for
amino-acid
--meta_data META_DATA, -md META_DATA
files contains the meta data
--metavar METAVAR, -mv METAVAR
name of the meta var (response variable)
--gap GAP, -gp GAP Threshold to drop positions that have GAPs above this
proportion. Default value is 0.7 and it means that the
positions that 70% or more GAPs will be dropped from
the analysis.
--miss_gap MISS_GAP, -mgp MISS_GAP
Threshold to impute missing values with GAP. Gapsin
positions that have missing values (gaps) above this
proportionare replaced with the term 'GAP'. the rest
of the missing valuesare replaced by the mode of each
position.
--ult_rare ULT_RARE, -u ULT_RARE
Threshold to modify the ultra rare cases in each
position.
--anatype {reg,cl}, -a {reg,cl}
type of analysis
--distance_metric {correlation,hamming,jaccard,normalized_mutual_info_score,adjusted_mutual_info_score,adjusted_rand_score}, -dm {correlation,hamming,jaccard,normalized_mutual_info_score,adjusted_mutual_info_score,adjusted_rand_score}
distance metric. Default is correlation.
--fraction FRACTION, -fr FRACTION
fraction of main data to run
--redundant_threshold REDUNDANT_THRESHOLD, -rt REDUNDANT_THRESHOLD
threshold for the p-value of the statistical tests to
drop redundant features. Defaultvalue is 0.25
--distance_threshold DISTANCE_THRESHOLD, -dth DISTANCE_THRESHOLD
threshold for the distance between positions to put
them in clusters. features with distances <= than the
threshold will be grouped together. Default values is
0.3
--top_models TOP_MODELS, -tm TOP_MODELS
number of top models to consider for merging the
results. Default value is 5
--aggregate AGGREGATE
the aggregate function for summarising the importance
values in thepositions. Can be a string representing a
built-in aggregation function (e.g., 'mean', 'max',
'min', 'std', etc.)
--cv CV, -cv CV number of folds for cross validation. Default is 10.
If the given number is less than 1, then instead of
CV, a train/test split approach will be used with cv
being the test size.
--test TEST, -t TEST test dataset ratio. A random sample of the main data
will be used as the test dataset. If size of test
dataset is less than 30 samples, then it will be
ignored. Default is 0.2.
--ref_id REF_ID, -r REF_ID
ID/order of the reference sequence in the sequence
file. Default is last sequence.
--ref_compare REF_COMPARE, -c REF_COMPARE
ID/order of the sequences to compare with the
reference sequence.
--compare_len COMPARE_LEN, -l COMPARE_LEN
length of output sequences
--tune After running the 10-fold cross validations, should
the top selected models be tuned and finalize, or
finalized only?
--plot plot all the individual positions that are
statistically significant.Depending on your data, this
process may produce many plots.
--write During reading the fasta file we delete the positions
that have GAPs over a certain threshold that can be
changed in the `gap_threshold` argumentin the
`read_data` function. As this may change the whole
FASTA file, you maywant to save the FASTA file after
this cleaning step.
--seed Seed for random number generator. Default is 123.
- correlated positions. We group all the colinear positions together.
- models summary. list of models and their performance metrics.
- plot of the feature importance of the top models in modelName_dpi.png format.
- csv files of feature importance based on top models containing, feature, importance, relative importance, group of the position (we group all the colinear positions together)
- plots and csv file of average of feature importance of top models.
- box plot (regression) or stacked bar plot (classification) for top positions of each model.
- pickle files of the plots and final models
- p-values of all the variables used in training of the final model
deepBreaks -sf PATH_TO_SEQUENCE.FASTA -st aa -md PATH_TO_META_DATA.tsv -mv
META_VARIABLE_NAME -a reg -dth 0.15 --plot --write
Multiple detailed jupyter notebook of deepBreaks implementation are available in the examples and the required data for the examples are also available in the data directory.
For the deepBreaks.models.model_compare function, these are the available models by default:
- Regression:
models = {
'rf': RandomForestRegressor(n_jobs=-1, random_state=123),
'Adaboost': AdaBoostRegressor(random_state=123),
'et': ExtraTreesRegressor(n_jobs=-1, random_state=123),
'gbc': GradientBoostingRegressor(random_state=123),
'dt': DecisionTreeRegressor(random_state=123),
'lr': LinearRegression(n_jobs=-1),
'Lasso': Lasso(random_state=123),
'LassoLars': LassoLars(random_state=123),
'BayesianRidge': BayesianRidge(),
'HubR': HuberRegressor(),
'xgb': XGBRegressor(n_jobs=-1, random_state=123),
'lgbm': LGBMRegressor(n_jobs=-1, random_state=123)
}- Classification:
models = {
'rf': RandomForestClassifier(n_jobs=-1, random_state=123),
'Adaboost': AdaBoostClassifier(random_state=123),
'et': ExtraTreesClassifier(n_jobs=-1, random_state=123),
'lg': LogisticRegression(n_jobs=-1, random_state=123),
'gbc': GradientBoostingClassifier(random_state=123),
'dt': DecisionTreeClassifier(random_state=123),
'xgb': XGBClassifier(n_jobs=-1, random_state=123),
'lgbm': LGBMClassifier(n_jobs=-1, random_state=123)
}The default metrics for evaluation are:
- Regression:
scores = {'R2': 'r2',
'MAE': 'neg_mean_absolute_error',
'MSE': 'neg_mean_squared_error',
'RMSE': 'neg_root_mean_squared_error',
'MAPE': 'neg_mean_absolute_percentage_error'
}- Classification:
scores = {'Accuracy': 'accuracy',
'AUC': 'roc_auc_ovr',
'F1': 'f1_macro',
'Recall': 'recall_macro',
'Precision': 'precision_macro'
}To get the ful list of available metrics, you can use:
from sklearn import metrics
print(metrics.SCORERS.keys())The default search parameters for the models are:
import numpy as np
params = {
'rf': {'rf__max_features': ["sqrt", "log2"]},
'Adaboost': {'Adaboost__learning_rate': np.linspace(0.001, 0.1, num=2),
'Adaboost__n_estimators': [100, 200]},
'gbc': {'gbc__max_depth': range(3, 6),
'gbc__max_features': ['sqrt', 'log2'],
'gbc__n_estimators': [200, 500, 800],
'gbc__learning_rate': np.linspace(0.001, 0.1, num=2)},
'et': {'et__max_depth': [4, 6, 8],
'et__n_estimators': [500, 1000]},
'dt': {'dt__max_depth': [4, 6, 8]},
'Lasso': {'Lasso__alpha': np.linspace(0.01, 100, num=5)},
'LassoLars': {'LassoLars__alpha': np.linspace(0.01, 100, num=5)}
}Attention: The names of models in the provided dict are the same with the names in the dict provided
for the params. If the name from the models dict does not match, the default sklearn parameters for that model
is then used. For example, model_compare_cv uses the xgboost with default hyperparameters.
To use the deepBreaks.models.model_compare_cv function with default parameters:
from deepBreaks.models import model_compare_cv
from deepBreaks.preprocessing import MisCare, ConstantCare, URareCare, CustomOneHotEncoder
from deepBreaks.preprocessing import FeatureSelection, CollinearCare
from deepBreaks.utils import get_models, get_scores, get_params, make_pipeline
ana_type = 'reg' # assume that we are running a regression analysis
report_dir = 'PATH/TO/A/DIRECTORY' # to save the reports
prep_pipeline = make_pipeline(cache_dir=None,
steps=[
('mc', MisCare(missing_threshold=0.25)),
('cc', ConstantCare()),
('ur', URareCare(threshold=0.05)),
('cc2', ConstantCare()),
('one_hot', CustomOneHotEncoder()),
('feature_selection', FeatureSelection(model_type=ana_type, alpha=0.25)),
('collinear_care', CollinearCare(dist_method='correlation', threshold=0.25))
])
report, top = model_compare_cv(X=tr, y=y, preprocess_pipe=prep_pipeline,
models_dict=get_models(ana_type=ana_type),
scoring=get_scores(ana_type=ana_type),
report_dir=report_dir,
cv=10, ana_type=ana_type, cache_dir=None)To use a new set of models, params, or metrics you can define them in a dict:
import deepBreaks.models as ml
from sklearn.ensemble import RandomForestRegressor, AdaBoostRegressor
from sklearn.ensemble import ExtraTreesRegressor
from deepBreaks.models import model_compare_cv
from deepBreaks.preprocessing import MisCare, ConstantCare, URareCare, CustomOneHotEncoder
from deepBreaks.preprocessing import FeatureSelection, CollinearCare
from deepBreaks.utils import get_models, get_scores, get_params, make_pipeline
ana_type = 'reg' # assume that we are running a regression analysis
report_dir = 'PATH/TO/A/DIRECTORY' # to save the reports
# define a new set of models
models = {'rf': RandomForestRegressor(n_jobs=-1, random_state=123),
'Adaboost': AdaBoostRegressor(random_state=123),
'et': ExtraTreesRegressor(n_jobs=-1, random_state=123)
}
prep_pipeline = make_pipeline(cache_dir=None,
steps=[
('mc', MisCare(missing_threshold=0.25)),
('cc', ConstantCare()),
('ur', URareCare(threshold=0.05)),
('cc2', ConstantCare()),
('one_hot', CustomOneHotEncoder()),
('feature_selection', FeatureSelection(model_type=ana_type, alpha=0.25)),
('collinear_care', CollinearCare(dist_method='correlation', threshold=0.25))
])
report, top = model_compare_cv(X=tr, y=y, preprocess_pipe=prep_pipeline,
models_dict=models,
scoring=get_scores(ana_type=ana_type),
report_dir=report_dir,
cv=10, ana_type=ana_type, cache_dir=None)
'''
Since we do not define a set of parameters for the model "et", it will fit with
default parameters
'''
# change the set of metrics
scores = {'R2': 'r2',
'MAE': 'neg_mean_absolute_error',
'MSE': 'neg_mean_squared_error'
}
report, top = model_compare_cv(X=tr, y=y, preprocess_pipe=prep_pipeline,
models_dict=models,
scoring=scores,
report_dir=report_dir,
cv=10, ana_type=ana_type, cache_dir=None)Here we try to use the deepBreaks on different datasets and elaborate on the results.

Opsins are genes involved in light sensitivity and vision, and when coupled with a light-reactive chromophore, the absorbance of the resulting photopigment dictates physiological phenotypes like color sensitivity. We analyzed the amino acid sequence of rod opsins because previously published mutagenesis work established mechanistic connections between 12 specific amino acid sites and phenotypes Yokoyama et al. (2008). Therefore, we hypothesized that machine learning approaches could predict known associations between amino acid sites and absorbance phenotypes. We identified opsins expressed in rod cells of vertebrates (mainly marine fishes) with absorption spectra measurements (λmax, the wavelength with the highest absorption). The dataset contains 175 samples of opsin sequences. We next applied deepBreaks on this dataset to find the most important sites contributing to the variations of λmax. This Jupyter Notebook illustrates the steps.
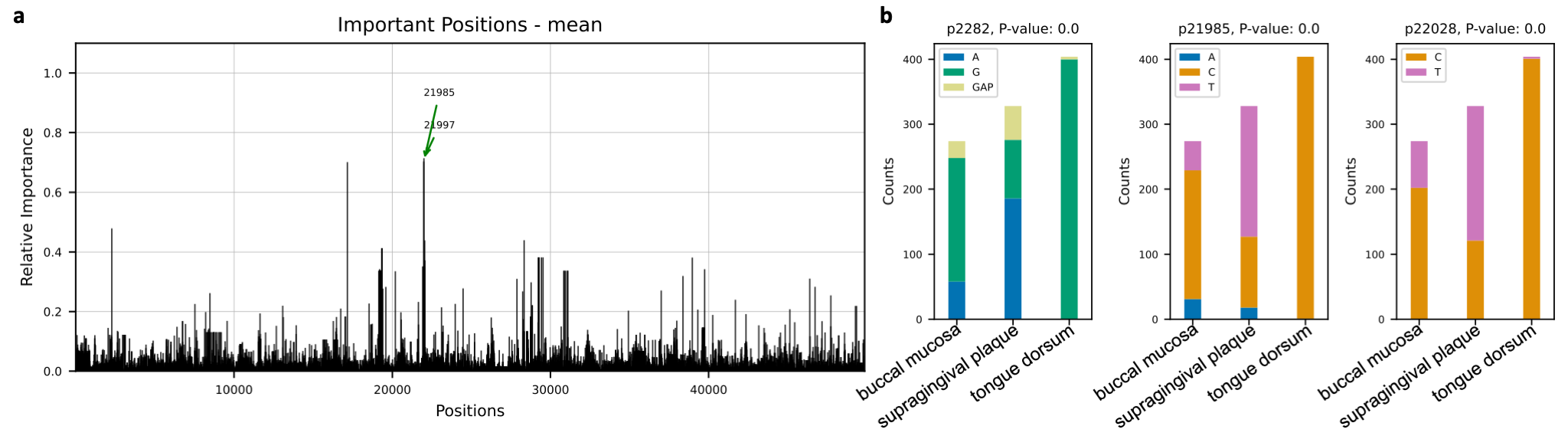
Microbial species tend to adapt at the genome level to the niche in which they live. We hypothesize
that genes with essential functions change based on where microbial species live. Here we use microbial strain
representatives from stool metagenomics data of healthy adults from the
Human Microbiome Project. The input for deepBreaks consists of 1) an MSA file
with 1006 rows, each a representative strain of a specific microbial species, here Haemophilus parainfluenzae, with
49839 lengths; and 2) labels for deepBreaks prediction are body sites from which samples were collected.
This Jupyter Notebook
illustrates the steps.
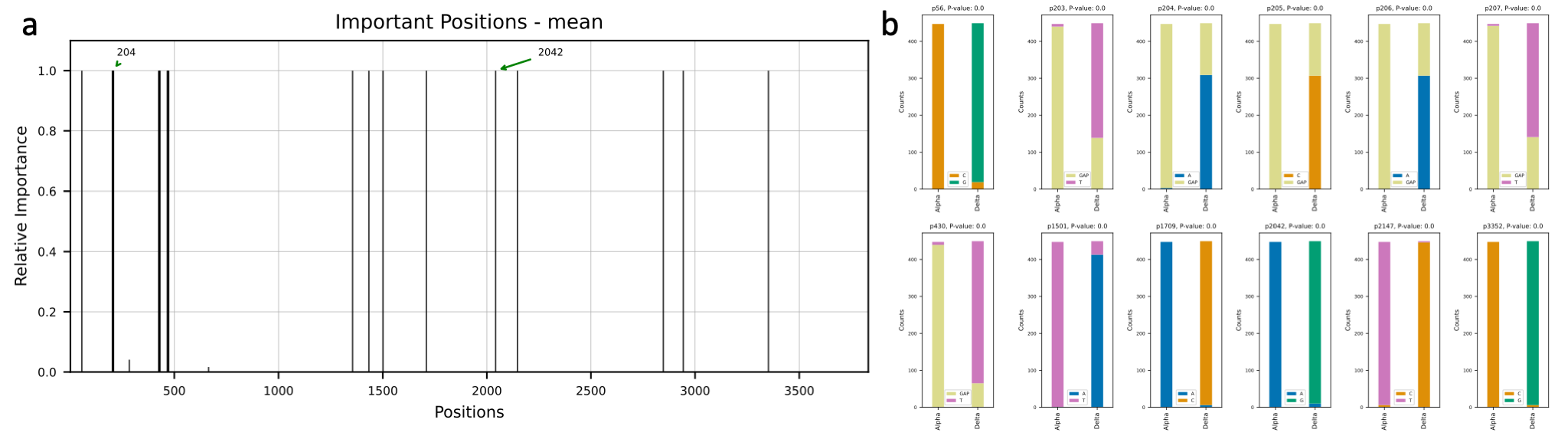 Variants occur with new mutations in the virus genome. Most mutations in the SARS-CoV-2 genome do not affect the
functioning of the virus. However, mutations in the spike protein of SARS-CoV-2, which binds to receptors on cells
lining the inside of the human nose, may make the virus easier to spread or affect how well vaccines protect people.
We are going to study the mutations in the spike protein of the sequences of Alpha (B.1.1.7): the first variant of
concern described in the United Kingdom (UK) in late December 2020 and Delta (B.1.617.2): first reported in India in
December 2020. We used the publicly available data from the GSAID and obtained 900 sequences
of spike protein region of Alpha (450 samples) and Delta (450 samples) variants. Then, we used deepBreaks to analyze
the data and find the most important (predictive) positions in these sequences in terms of classifying the variants.
This
Jupyter Notebook
illustrates the steps.
Variants occur with new mutations in the virus genome. Most mutations in the SARS-CoV-2 genome do not affect the
functioning of the virus. However, mutations in the spike protein of SARS-CoV-2, which binds to receptors on cells
lining the inside of the human nose, may make the virus easier to spread or affect how well vaccines protect people.
We are going to study the mutations in the spike protein of the sequences of Alpha (B.1.1.7): the first variant of
concern described in the United Kingdom (UK) in late December 2020 and Delta (B.1.617.2): first reported in India in
December 2020. We used the publicly available data from the GSAID and obtained 900 sequences
of spike protein region of Alpha (450 samples) and Delta (450 samples) variants. Then, we used deepBreaks to analyze
the data and find the most important (predictive) positions in these sequences in terms of classifying the variants.
This
Jupyter Notebook
illustrates the steps.
 Subtypes of the human immunodeficiency virus type 1 (HIV-1) group M are different in the envelope (Env) glycoproteins
of the virus. These parts of the virus are displayed on the surface of the virion and are targets for both neutralizing
antibody and cell-mediated immune responses. The third hypervariable domain (V3) of HIV-1 gp120 is a cysteine-bounded
loop structure usually composed of 105 nucleotides and labeled as the base (nu 1:26 and 75:105), stem
(nu 27:44 and 54:74), and turn (nu 45:53) regions Lynch et al. (2009) .
Among all of the hyper-variable regions in gp120 (V1-V5), V3 is playing the main role in the virus infectivity
Felsövályi et al. (2006).
Here we useare using deepBreaks to identify important regions in the V3 loop that are important in terms of associating
the V3 sequences V3 to subtypes B and C. We used the Los Alamos HIV Database to gather the
nucleotide sequences of the V3 loop of subtypes B and C.
This Jupyter Notebook
illustrates the steps.
Subtypes of the human immunodeficiency virus type 1 (HIV-1) group M are different in the envelope (Env) glycoproteins
of the virus. These parts of the virus are displayed on the surface of the virion and are targets for both neutralizing
antibody and cell-mediated immune responses. The third hypervariable domain (V3) of HIV-1 gp120 is a cysteine-bounded
loop structure usually composed of 105 nucleotides and labeled as the base (nu 1:26 and 75:105), stem
(nu 27:44 and 54:74), and turn (nu 45:53) regions Lynch et al. (2009) .
Among all of the hyper-variable regions in gp120 (V1-V5), V3 is playing the main role in the virus infectivity
Felsövályi et al. (2006).
Here we useare using deepBreaks to identify important regions in the V3 loop that are important in terms of associating
the V3 sequences V3 to subtypes B and C. We used the Los Alamos HIV Database to gather the
nucleotide sequences of the V3 loop of subtypes B and C.
This Jupyter Notebook
illustrates the steps.
Here are the types of plots that are generated by deepBreaks to help you interpret the results.
- Feature importance plot: This plot shows the importance of each position in the sequences. The importance is calculated based on the feature importance of the top models. For each of the top models, the feature importance is created and also a plot for the average importance of the positions based on all the top models is created. The height of the bars in the plot shows the importance of the positions. The x-axis shows the positions in the sequences based on the raw input file.
- Box plot (regression) or stacked bar plot (classification): This plot shows the changes in the response variable based on the top positions. The x-axis shows the values in the positions and the y-axis shows the response variable (regression) or count of observations (classification).

- Scatter plot (regression) or confusion matrix (classification): This plot shows the relationship between the
response variable and the predicted values. This plot only created when the
--testargument is used.

One of the outputs of deepBreaks is a text file containing positions that are crucial for predicting the response
variable. This file, named imp_seq.txt, highlights the segments of the sequence with these important positions (above).
You can use these sequences to run BLAST and determine the functional properties of these positions or identify which gene they belong to. An example provided in the manuscript involves SARS-CoV-2 sequences. By using the reference genome available in the NCBI database (NC_045512.2), we can analyze the functional properties or mutations at these significant positions.
Here is a demonstration of the steps:

- Please submit your questions or issues with the software at Issues tracker.
- For community discussions, questions, and issue reporting, please visit our forum here