Home
This page describes the concept and architecture of the Cellxgene Gateway
The Cellxgene project from the Chan Zuckberg Institute allows rich visualization of single cell RNA seq data. However, it is limited to visualizing a single dataset at a time. This repo contains Cellxgene Gateway, a small python/flask app that allows you to host an unlimited number of datasets on a single server. It dynamically launches instances of Cellxgene Server, and spins them down after a period of inactivity.
The Gateway mediates between the incoming request, which always passes through a fixed domain name and port, and multiple cellxgene servers (one per dataset) that are either running in separate processes on a single server (currently implemented) or on an external docker container (potential improvement).
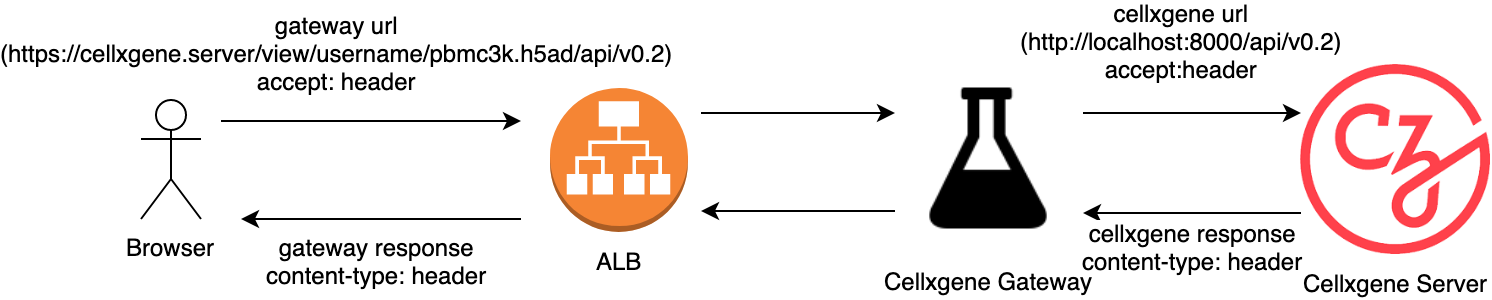
The role of the cellxgene gateway is
- translate incoming requests that mention the dns name and port of the ALB into requests for the cellxgene server running in the VPC.
- It must preserve the incoming accept header.
- translate outgoing responses that mention the cellxgene basepath into responses that mention the gateway base path.
- It must preserve the outgoing content-type header.
- It must translate the protocol for external resources to be consistent with the gateway basepath to avoid mixed content errors
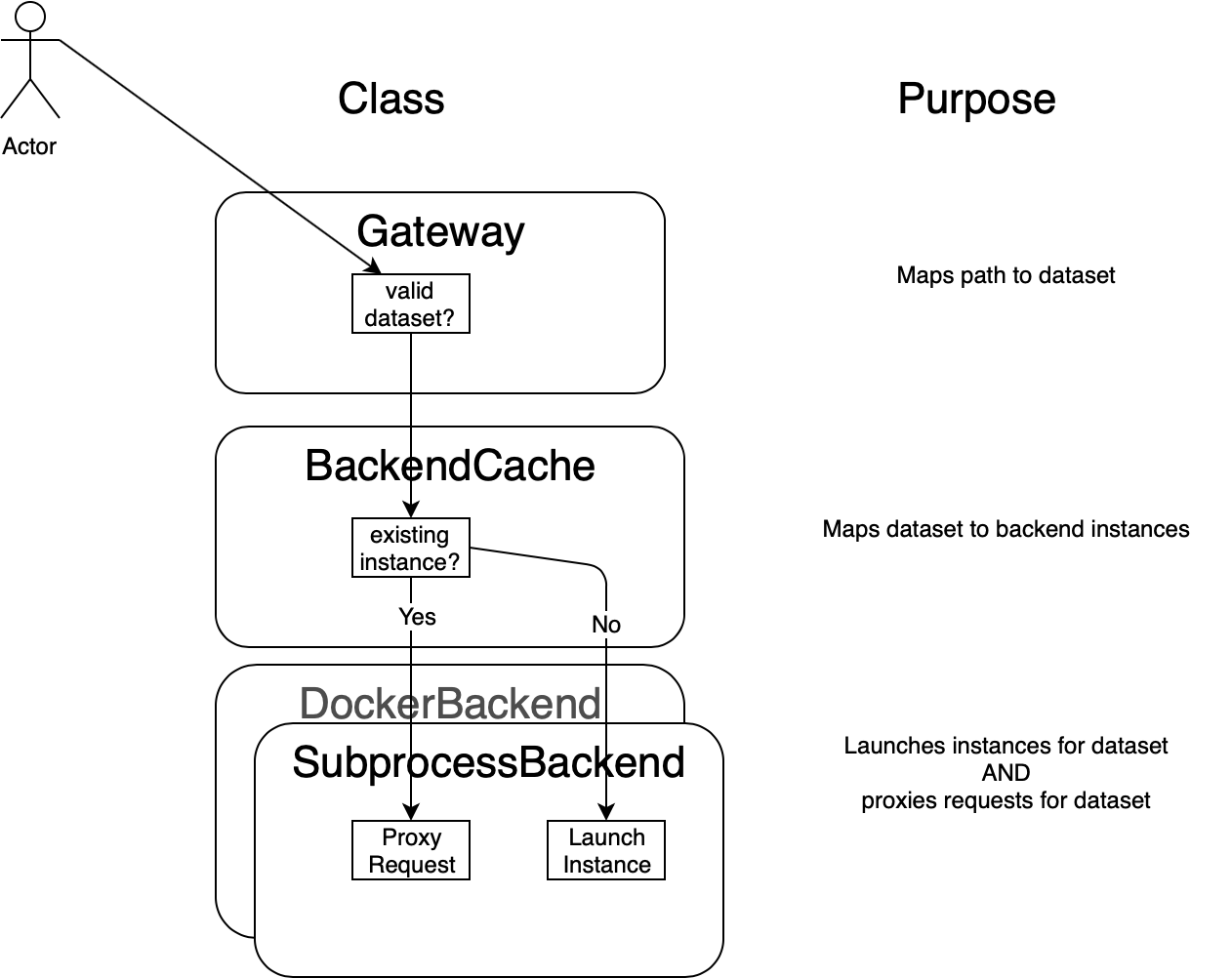
The basic idea here is to write a http://flask.pocoo.org/ app that receives all requests for cellxgene.server. It will fork a process running cellxgene for each dataset, and keep track of which processes are running by creating a file in /tmp/cellxgene-instances. Although this approach will not scale beyond a few concurrent datasets, it is easier to implement than the docker container approach and has significant overlap, so it is a reasonably first step.
Instances are tracked in the pid_list array within the PidCache class. Each element of the array has the following structure:
{
dataset: '/username/hpc/P11Merged_noCC',
pid: 31245,
port: 8101,
last_access: <unix timestamp>
}
Where
-
datasetis a path fragment identifying dataset. This is used to identify when we have an existing cellxgene process for a dataset. -
pidis the process id of the spawned process running cellxgene. This is used to kill the process when the dataset has been idle for too long. -
portis the port that the spawned process is listening on. This is used for the forwarded http requests. -
last_accessis the last time a request for any resource was served for the dataset
The actual data files are located at cellxgene_efs + dataset.
- If there is no path provided,
- provide an index of the available datasets with links that include the path.
- the index can be produced by iterating over all the directories/files similar to SPRING
- Else verify the path provided corresponds to an existing dataset
- Typical full url would be https://spring.server/username/hpc/P11Merged_noCC
- If there is no matching dataset, return a "not found" message
- Load all of the files from /tmp/cellxgene-instances
- If there is no file with matching dataset
- Grab lock for dataset with e.g. shared_lock = rwlock.SharedLock('/username/hpc/P11Merged_noCC')
- select an unused port. We can simply start from 8100 and increment until we find an unused port.
- Spawn cellxgene to display that dataset on the specified port
- Write .txt
- Release the dataset lock
- Forward request to cellxgene, send response back to client
Here is the general form of requests that the browser will send to the flask app, which we have called "cellxgene gateway":
https://cellxgene.server/view/<dataset><subpath>
// example:
https://cellxgene.server/view/username/pbmc3k.h5ad/api/v0.2
When running the gateway locally on port 5000, the browser side would be http://localhost:5000/view/username/pbmc3k.h5ad/api/v0.2
In both cases, we will call the part before the subpath (including the dataset) the "gateway basepath". Note two things
- The gateway basepath NEVER ends in a slash.
- The subpath ALWAYS begins with a slash.
- The dataset will always match the path to a dataset on disk, relative to the dataset root directory.
- get the port from the dataset name, in the example 'username/pbmc3k.h5ad'
- Make a request to the cellxgene server at http://localhost:. We will call the part without the subpath the "cellxgene basepath"
- Make sure to copy all request headers from the incoming "gateway request" to the outgoing "cellxgene request"
- Convert the "cellxgene response" that comes back into a "gateway response" that can be sent to the browser
- Replace all instances of the cellxgene basepath with the gateway basepath
- copy the relevant response headers, including "Content-Type" from the cellxgene response to the flask response
- return flask response to the browser
| Part | Example |
|---|---|
| gateway url | https://cellxgene.server/view/username/pbmc3k.h5ad/api/v0.2 |
| gateway basepath | https://cellxgene.server/view/username/pbmc3k.h5ad |
| gateway host | cellxgene.server (localhost:5005 when running locally) |
| path | /username/pbmc3k.h5ad/api/v0.2 |
| dataset | username/pbmc3k.h5ad |
| subpath | /api/v0.2 |
| cellxgene basepath | http://localhost:8000 |
| cellxgene url | http://localhost:8000/api/v0.2 |
In theory, to support Docker we need the following changes:
- Instead of spawning a cellxgene process, we need to launch a docker container that can mount EFS and display the dataset
- The .txt files should become docker information files named after some container identifier provided by AWS
- The files should be stored on the cellxgene EFS (shared filesystem) instead of in /tmp.
I'll let you know how it goes in practice if we ever get to it 😄 .
In addition to the Docker backend, there are a couple things that would be nice to clean up if this continues to be used,
- The current implementation expects all datasets to be available on a mounted file system. It would be nice to allow it to use files from S3 or other kinds of file/object stores.
- Customizing the look and feel of the front end is rather messy at the moment. Adding a mechanism to override the Jinja templates may help.