
Detect and visualize target mutations by scanning FastQ files directly
- Features
- Application scenarios
- Take a quick glance
- Download, compile and install
- HTML report
- JSON report
- All options
- Customize your mutation file
- Work with BAM/CRAM
- Remarks
- Cite MutScan
- Ultra sensitive, guarantee that all reads supporting the mutations will be detected
- Can be 50X+ faster than normal pipeline (i.e. BWA + Samtools + GATK/VarScan/Mutect).
- Very easy to use and need nothing else. No alignment, no reference genome, no variant call, no...
- Contains built-in most actionable mutation points for cancer-related mutations, like EGFR p.L858R, BRAF p.V600E...
- Beautiful and informative HTML report with informative pileup visualization.
- Multi-threading support.
- Supports both single-end and pair-end data.
- For pair-end data, MutScan will try to merge each pair, and do quality adjustment and error correction.
- Able to scan the mutations in a VCF file, which can be used to visualize called variants.
- Can be used to filter false-positive mutations. i.e. MutScan can handle highly repetive sequence to avoid false INDEL calling.
- you are interested in some certain mutations (like cancer drugable mutations), and want to check whether the given FastQ files contain them.
- you have no enough confidence with the mutations called by your pipeline, so you want to visualize and validate them to avoid false positive calling.
- you worry that your pipeline uses too strict filtering and may cause some false negative, so you want to check that in a fast way.
- you want to visualize the called mutation and take a screenshot with its clear pipeup information.
- you called a lot of INDEL mutations, and you worry that mainly they are false positives (especially in highly repetive region)
- you want to validate and visualize every record in the VCF called by your pipeline.
- ...
- Sample HTML report: http://opengene.org/MutScan/report.html
- Sample JSON report: http://opengene.org/MutScan/report.json
- Dataset for testing: http://opengene.org/dataset.html
- Command to test
mutscan -1 R1.fq.gz -2 R2.fq.gzconda install -c bioconda mutscanThis binary is only for Linux systems: http://opengene.org/MutScan/mutscan
# this binary was compiled on CentOS, and tested on CentOS/Ubuntu
wget http://opengene.org/MutScan/mutscan
chmod a+x ./mutscan# get source (you can also use browser to download from master or releases)
git clone https://github.com/OpenGene/MutScan.git
# build
cd mutscan
make
# Install
sudo make installIf you want to compile MutScan on Windows, you should use cygwin. We already built one with cygwin-2.6.0/g++ 5.4, and it can be downloaded from:
http://opengene.org/MutScan/windows_mutscan.zip
- A HTML report will be generated, and written to the given filename. See http://opengene.org/MutScan/report.html for an example.
- If you run the command in your Linux server and want to view the HTML report on your local system. DO remember to copy all of the
xxxx.htmlandxxxx.html.filesand keep them in the same folder, then clickxxxx.htmlto view it in browser. - The default file name is
mutscan.html, and a foldermutscan.html.fileswill be also generated. - By default, an indivudal HTML file will be generated for each found mutation. But you can specify
-sor--standaloneto contain all mutations in a single HTML file. Be caution with this mode if you are scanning too many records (for example, scanning VCF), it will give you a very big HTML file and is not loadable by browser. - Here is a screenshot for the pileup of a mutation (EGFR p.T790M) generated by MutScan:
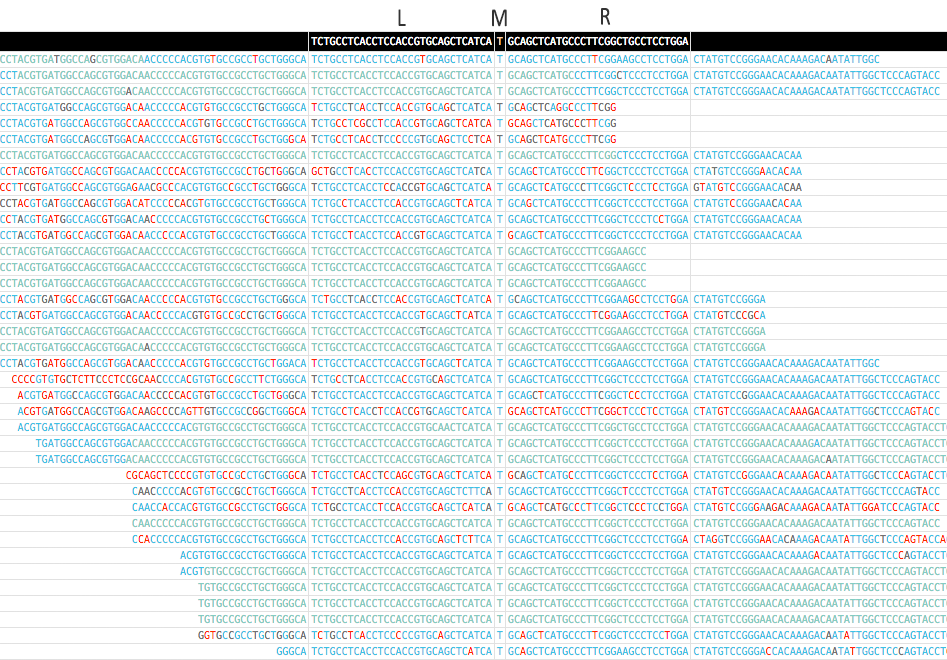
- An pileup of EGFR p.T790M mutation is displayed above. EGFR p.T790M is a very important drugable mutation for lung cancer.
- The color of each base indicates its quality, and the quality will be shown when mouse over.
- In first column, d means the edit distance of match, and --> means forward, <-- means reverse
JSON report is disabled by default. You can enable it by specifying a JSON file name using -j or --json. A JSON report is like this:
{
"command":"./mutscan -1 /Users/shifu/data/fq/S010_20170320003-4_ffpedna_pan-cancer-v1_S10_R1_001.fastq -2 /Users/shifu/data/fq/S010_20170320003-4_ffpedna_pan-cancer-v1_S10_R2_001.fastq -h z.html -j z.json -v --simplified=off ",
"version":"1.14.0",
"time":"2018-05-15 15:48:21",
"mutations":{
"NRAS-neg-1-115258747-2-c.35G>C-p.G12A-COSM565":{
"chr":"chr1",
"ref":["TGGATTGTCAGTGCGCTTTTCCCAACACCA","G","CTGCTCCAACCACCACCAGTTTGTACTCAG"],
"reads":[
{
"breaks":[31,61,62,76],
"seq":"ATATTCATCTACAAAGTGGTTCTGGATTAGCTGGATTGTCAGTGCGCTTTTCCCAACACCAGCTGCTCCAACCACC",
"qual":"eeeeeiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiieiiiiiiiiiiieieeeee"
},
{
"breaks":[31,61,62,76],
"seq":"ATATTCATCTACAAAGTGGTTCTGGATTAGCTGGATTGTCAGTGCGCTTTTCCCAACACCAGCTGCTCCAACCACC",
"qual":"eeeeeiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiieeeee"
}
]
},
"PIK3CA-pos-3-178936082-9-c.1624G>A-E542K-COSM760":{
"chr":"chr3",
"ref":["AAAGCAATTTCTACACGAGATCCTCTCTCT","A","AAATCACTGAGCAGGAGAAAGATTTTCTAT"],
"reads":[
{
"breaks":[22,52,53,83],
"seq":"GGAAAATGACAAAGAACAGCTCAAAGCAATTTCTACACGAGATCCTCTCTCTAAAATCACTGAGCAGGAGAAAGATTTTCCAAAGATGTTTCTCAGAACGCTGCAGTCTGCAATTTGTATGAATTCCC",
"qual":"eeeeeiiiQiiiiiieiiiieiSeiiiiiie`iiii`i`iiiiiiiiiiiiii`iiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiaiiiiiiiiiiiiiiiiiieiiiiiieeeee"
},
{
"breaks":[0,27,28,58],
"seq":"GCAATTTCTACACGAGATCCTCTCTCTAAAATCACTGCGCAGGAGAAAGATTTTCTATGGACCACAGGTAAGTGCTAAAATGGAGATTCTCTGTTTCTTTTTCTTTATTACAGAAAAAATAACTGACTTTGGCTGATCTCAGCATGTTTTTACCATACC",
"qual":"AAAAAEEEEiieiiieiiiiiiiiiieiiiiiiiie``iiiiiieiiiiiiiiiieiiiieiieieeiiiSiiiiiieiiiiiiiiiiiiiieiiiiiSiiiiiiiiiiiiieiiiiiiiiiiii`ieiiieiii`ieiiiii`eS``eieEEEAAAAA"
}
]
}
}
}usage: mutscan -1 <read1_file> -2 <read2_file> [options]...
options:
-1, --read1 read1 file name, required
-2, --read2 read2 file name
-m, --mutation mutation file name, can be a CSV format or a VCF format
-r, --ref reference fasta file name (only needed when mutation file is a VCF)
-h, --html filename of html report, default is mutscan.html in work directory
-j, --json filename of JSON report, default is no JSON report (string [=])
-t, --thread worker thread number, default is 4
-S, --support min read support required to report a mutation, default is 2.
-k, --mark when mutation file is a vcf file, --mark means only process the records with FILTER column is M
-l, --legacy use legacy mode, usually much slower but may be able to find a little more reads in certain case
-s, --standalone output standalone HTML report with single file. Don't use this option when scanning too many target mutations (i.e. >1000 mutations)
-n, --no-original-reads dont output original reads in HTML and text output. Will make HTML report files a bit smaller
-?, --help print this messageThe plain text result, which contains the detected mutations and their support reads, will be printed directly. You can use > to redirect output to a file, like:
mutscan -1 <read1_file_name> -2 <read2_file_name> > result.txtMutScan generate a very informative HTML file report, default is mutscan.html in the work directory. You can change the file name with -h argument, like:
mutscan -1 <read1_file_name> -2 <read2_file_name> -h report.html
For single-end sequencing data, -2 argument is omitted:
mutscan -1 <read1_file_name>
-t argument specify how many worker threads will be launched. The default thread number is 4. Suggest to use a number less than the CPU cores of your system.
- Mutation file, specified by
-m, can be aCSV file, or aVCF file. - If no
-mspecified, MutScan will use the built-in default mutation file with about 60 cancer related mutation points. - If a CSV is provided, no reference genome assembly needed.
- If a VCF is provided, corresponding reference genome assembly should be provided (i.e. ucsc.hg19.fasta), and should not be zipped.
A CSV file with columns of name, left_seq_of_mutation_point, mutation_seq, right_seq_of_mutation_point and chromosome(optional)
#name, left_seq_of_mutation_point, mutation_seq, right_seq_of_mutation_point, chromosome
NRAS-neg-1-115258748-2-c.34G>A-p.G12S-COSM563, GGATTGTCAGTGCGCTTTTCCCAACACCAC, T, TGCTCCAACCACCACCAGTTTGTACTCAGT, chr1
NRAS-neg-1-115252203-2-c.437C>T-p.A146V-COSM4170228, TGAAAGCTGTACCATACCTGTCTGGTCTTG, A, CTGAGGTTTCAATGAATGGAATCCCGTAAC, chr1
BRAF-neg-7-140453136-15-c.1799T>A -V600E-COSM476, AACTGATGGGACCCACTCCATCGAGATTTC, T, CTGTAGCTAGACCAAAATCACCTATTTTTA, chr7
EGFR-pos-7-55241677-18-c.2125G>A-p.E709K-COSM12988, CCCAACCAAGCTCTCTTGAGGATCTTGAAG, A, AAACTGAATTCAAAAAGATCAAAGTGCTGG, chr7
EGFR-pos-7-55241707-18-c.2155G>A-p.G719S-COSM6252, GAAACTGAATTCAAAAAGATCAAAGTGCTG, A, GCTCCGGTGCGTTCGGCACGGTGTATAAGG, chr7
EGFR-pos-7-55241707-18-c.2155G>T-p.G719C-COSM6253, GAAACTGAATTCAAAAAGATCAAAGTGCTG, T, GCTCCGGTGCGTTCGGCACGGTGTATAAGG, chr7
testdata/mutations.csv gives an example of CSV-format mutation file
A standard VCF can be used as a mutation file, with file extension .vcf or .VCF. If the mutation file is a VCF file, you should specify the reference assembly file by -r <ref.fa>. For example the command can be:
mutscan -1 R1.fq -2 R2.fq -m target.vcf -r hg19.faIf you want to run MutScan with BAM/CRAM files, you can use samtools to convert them to FASTQ files using samtools fastq command, both single-end and paired-end data are supported by latest version of samtools fastq.
MutScanrequires at least 50 bp long reads, if your reads are too short, do not use it- If you want to extract mutations even with only one read support, add
-S 1or--support=1in the command - Feel free to raise an issue if you meet any problem
Shifu Chen, Tanxiao Huang, TieXiang Wen, Hong Li, Mingyan Xu and Jia Gu. MutScan: fast detection and visualization of target mutations by scanning FASTQ data. BMC Bioinformatics. https://doi.org/10.1186/s12859-018-2024-6