the practice of bioinformatics concepts 🧬🔬💻
Def: bioinformatics is a subdiscipline of biology and computer science concerned with the acquisition, storage, analysis, and dissemination of biological data, most often DNA and amino acid sequences.
Find the reverse complement of a DNA string - the complementary strand.
input: a DNA string s of length at most 1000 bp (base pairs).
ouput: the reverse complement sc of s.
For example: the string AAAACCCGGT has a reverse complement string ACCGGGTTTT
Identify unknown DNA quickly by computing the GC-content of a DNA string.
input: at most 10 strings of DNA in FASTA format (of length at most 1kbp)
ouput: the ID of the string having the highest GC-content, the GC-content of that string within 0.001 absolute error.
Fasta Format: A text format used for naming genetic strings in databases. See link directly below.
With a collection of k-mers, form an adjacency list (that represents a De-Buijn graph). This list can be used to reconstruct possible DNA strings.
input: a collection of k-mers Patterns
ouput: the de-bruijn graph, in the form of an adjacency list (a list that shows directly adjacent-connected nodes, which are overlapping 3-mers)
Here, prefixes -> suffixes (for kmer GAGG, prefix GAG -> suffix AGG, because AGG exists in our suffixes) and (for kmer CAGG, prefix CAG -> AGG, AGG again, becuase CAGG appears twice in our collection.)
De-bruijn Graph: a graph to show the connectedness of the kmers in the input collection. ref
With a set kmer length, determine the constructed debruijn graph, in the form of an adjacency list, from a DNA string.
input: an integer k and a genome DNA string
ouput: a de-bruijn graph (with k overlap), in the form of an adjacency list (a list that shows the overlapping, directly adjacent-connected nodes, a list containing the edges of the graph)
For example: if the integer k is 4, the resulting adj-list will show (Node -> Node) like (AAG -> AGA), where the rightmost AG of the first node overlaps with the leftmost AG from the second node
With a collection of Patterns of k-mers, create an overlap graph in the form of an adjacency list. The length k is the length of any of the kmers in the input collection.
input: a collection Patterns of k-mers
ouput: the overlap graph from the given patterns, in the form of an adjacency list
Overlap Graph: Here, the overlap graph is constructed dependending on which kmer nodes are overlapping, hence the name. The overlapping length of the kmer is k-1. For exmaple: kmer CATGC -> ATGCG, because the ATGC from the right side of the first kmer node overlaps with the ATGC from the left side of the second kmer node.
Determine the count of nucleobases in a string of DNA. ("A", "C", "G", "T" in a string of DNA). The beginnings of quantitative analysis for the genome, and several strings of DNA.
input: a DNA string s of length at most 1000 nt (nucleotides)
output: four integers (sep. by spaces) counting the respective number of times that the symbols "A", "C", "G", "T" occur in s.
Determine the number of mutations between two strings of DNA. The difference of these two strings is an integer, also known as the Hamming distance.
input: two DNA strings s and t of equal length (not exceding 1 kbp)
output: the Hamming distance dH(s, t), or the number of different bases (that results from comparing both strings)
The main goal of this project was to determine how we can sequence the human genome for the purpose of creating effective antibiotics. In this project we looked at a brief history of antibiotics, we examined their use and overall effectiveness (and forthcoming ineffectiveness, that is, with antibiotic-resistant bacteria), proteomics' role in the the development of medicines, and finally, we broke down the Central Dogma of Molecular Biology to computationally accomplish our goals. And to the last point, when we understand "DNA makes RNA makes protein", we can break that process down further to fully understand how we can create effective antibiotics.
The first part: Translate an RNA string into an amino acid string. By finding the amino acid sequences, we are able to find certain antibiotics as well as make profound discoveries that relate to amino acids, including how Bacteria or other organisms produce such antibiotics, or other products beneficial to humans.
The second part: Find substrings of a genome that encodes for a given amino acid string. A, sort of, reverse engineering process that can determine where in a string of DNA a certain amino acid string is encoded.
Please see the final report below for a better explanation of this project.
-
There are three different ways to divide a DNA string into codons for translation, one starting at each of the girst three starting positions of the string. Also, a DNA string Pattern encodes an amino acid string Peptide if the RNA string transcribed from either Pattern of its reverse complement Pattern translates into Peptide. The initial three divisions * (the reverse complement pattern + regular pattern) = 6 strings to look at.
input:a DNA string and an amino acid string Peptideoutput:all substrings of the DNA string encoding Peptide (if they exist)For example: the DNA string "ATGGCCATGGCCCCCAGAACTGAGATCAATAGTACCCGTATTAACGGGTGA" and the peptide "MA" will output these sections of DNA -> ATGGCC, GGCCAT, and ATGGCC.
Reading frames: the different ways of dividing a DNA string into codons. Since DNA is double-stranded, a genome has six reading frames (three from each strand).
-
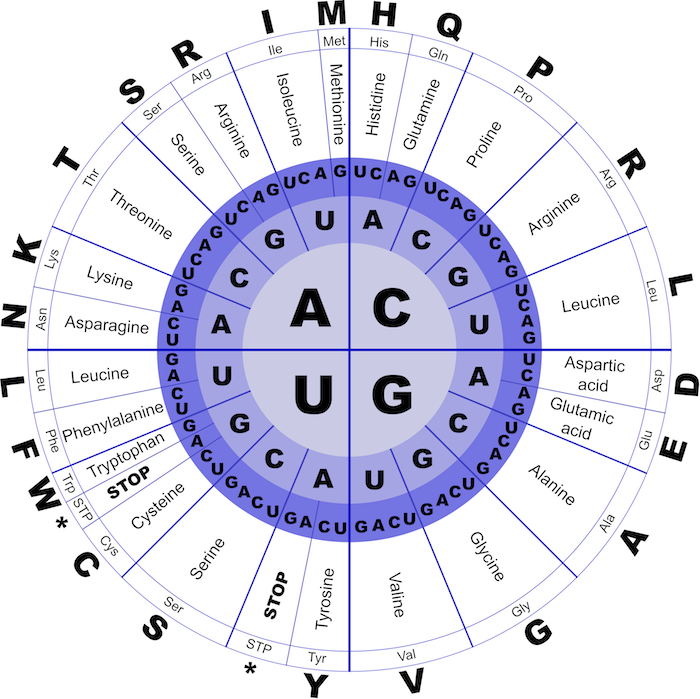
Normally coming from the transcription process where DNA is converted into RNA, we now have the RNA go through translation, where the mRNA is converted into a peptide chain for the creation of a protein. The process of translation converts each 3-mer (a set of three bases called a codon), into one of 20 amino acids. ref
input:an RNA string Patternoutput:the translation of Pattern into an amino acid string PeptideFor example: the pattern "AUGGCCAUGGCGCCCAGAACUGAGAUCAAUAGUACCCGUAUUAACGGGUGA" translates to the amino acid string "MAMAPRTEINSTRING"
{kind=link}
At what indexes does a substring of DNA exist in a second, longer piece of DNA. A substring is a contiguous collection of symbols that is contained in a larger string.
input: two DNA strings s and t (each of length at most 1 kbp)
output: all locations (starting indexes) of t as a substring of s
Find the k-length pieces of DNA found in a larger piece of DNA. The collection of these smaller pieces of DNA is known as the k-mer composition (of that string of DNA).
input: an integer k and a string of DNA
output: a Compositionk(DNA string) (the k-mers, which can be returned in any order)
For example: if k = 5 and the DNA string = "CAATCCAAC", then the output composition would be these strings of DNA -> {AATCC, ATCCA, CAATC, CCAAC, TCCAA}.
Piece together a collection of kmers that are in order of the final genome path.
input: a sequence of k-mers
ouput: a DNA string where the k-mers have overlapped
For example: the input kmers in this specific order, ACCGA, CCGAA, CGAAG, GAAGC, AAGCT, form the resulting DNA string ACCGAAGCT. Here, the CCGA from the right side of the first kmer overlaps with the CCGA from the left side of the second kmer. Each subsequent kmer overlaps with the following kmer in a similar fashion to form the final DNA string.
Transcription requires the replacement of all instances of "T" in the initial DNA string t with "U", to form the resulting RNA string of t.
input: a DNA string t having length at most 1000 nt (a monomer making up a nucleic acid)
ouput: the transcribed RNA string of t