Reference genomes have become central to bioinformatics approaches, and form the core of standard analyses using contemporary sequencing data. However, the use of linear reference genomes, which provide the sequence of one representative genome for a species, is increasingly becoming a limitation as the number of sequenced genomes grows. In particular, they tend to bias us away from the observation of variation in the genomes we study. A general solution to this problem is to use a pangenome that incorporates both sequence and variation from many individuals as our reference system. This pangenome is naturally modeled as a graph with annotations, and can provide all the functionality traditionally provided by linear reference genomes. Unlike linear reference genomes, a pangenome readily incorporates both small and large variation, allowing bias-free genotyping at known alleles. In this course we will explore the use of modern bioinformatic tools that allow researchers to use pangenomes as their reference system when engaging in studies of organisms of all types. Such techniques will aid any researcher working on organisms of high genetic diversity or on organisms lacking a high-quality reference genome. This course targets all researchers interested in learning about an exciting paradigm shift in computational genomics.
This course is oriented towards biologists and bioinformaticians with at least an intermediate level of experience working with sequencing data formats and methods in the unix shell. The course will be of particular interest to researchers investigating organisms without a reference genome or populations featuring high levels of genetic diversity.
Practical 1: toy examples
Practical 2: HIV
Practical 3: Bacteria
Practical 4: MHC
Participants first will learn about limitations of linear reference-based methods and work through a brief refresher or introduction to standard approaches for processing sequencing data, including read alignment and variant calling. Provided these motivating examples, we will use data from a variety of relevant sources to develop an intuition about pangenomic methods and a practical familiarity with applicable tools.
We will mostly use vg and other methods that read and write GFA.
For our exploration we have collected several data sets that combine genomic and phenotypic information, with a focus on contexts that are difficult to approach using standard linear reference methods:
- Bacteria (with resistance phenotypes) http://www.nature.com/articles/nmicrobiol201641
- HLA typing, e.g. from ONT data (tobi can ask whether we can use data from Wigard Klosterman)
- Yeast (with growth phenotypes) NCYC collection TODO Erik: get data from Ignacio
- Viral pooled population studies (HPV?)
Using these data we work through various modules based on different functional aspects of pangenomics.
This figure, from a recent paper on computational pangenomics, can serve as a helpful guide to our learning objectives:
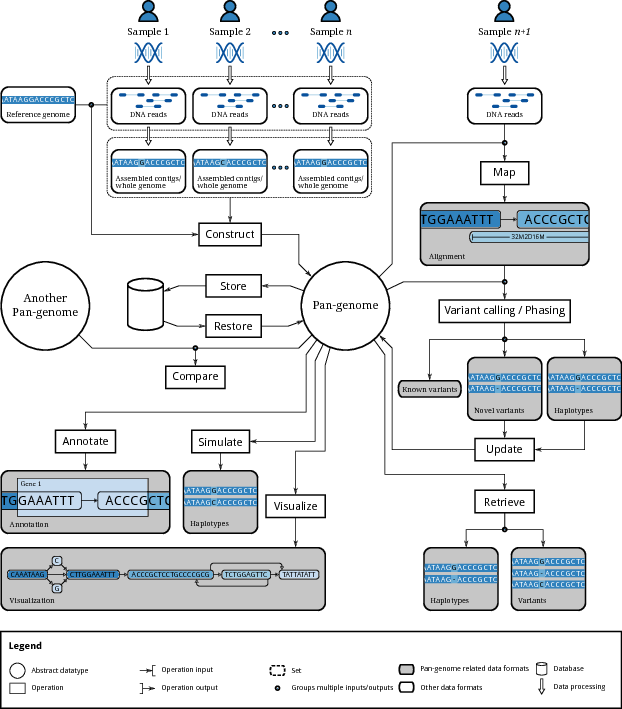
Our goal is that students achieve fluency in the practical aspects of relevant workflows within this scope.
Objectives:
- Know what a FASTA reference is, what a VCF or variant list is.
- Understand when we cannot simply align reads to a single reference and hope to correctly infer the unknown genome from which these reads are derived.
- Appreciate standard approaches to mitigate this issue.
Exercises:
- Align reads to a linear reference using bwa mem and call variants using freebayes.
- Align reads from a structural variant containing locus to a linear reference, visualize with IGV, and report what is found.
- Attempt to call variants in the region of a structural variant treated this way and discuss the results.
Objectives:
- Be able to describe a simple graphical model used for pangenome representation (vg's data model).
- Understand the relationship between traditional lists of variants, linear references, and basic pangenomic models.
- Have a basic understanding of the graphical models used by assemblers and how they relate to the data model.
Exercises:
- Generate a short paragraph summarizing the vg schema (using the schema itself, help from the instructors, and online documentation).
- Build a vg graph for a fragment of the human 1000 Genomes Project data using
vg construct.
Exercises:
- Run an assembler on a trivial (but real) data set and visualize the result with Bandage. (TODO: which data?)
- Write a script to count the contigs and edges in a GFA format output from this assembly process.
Exercises:
- Apply
vg msgaandcactusto assemble a graph from a fragment of the human MHC.
Objectives:
- Know basic techniques to visualize the sometimes-unweily pangenome systems.
Exercises:
Take graphs generated in our previous exercises and visualize them with:
- Bandage
- vg view
- IVG (we'll return to this later when we do alignments)
Objectives:
- Understand the basic principles behind read alignment, and how these are complicated by aligning reads to a graph.
- Have the ability to make alignments and interrogate the result to learn basic information like read coverage or putative variants.
Exercises:
- TODO
TODO

Tobias Marschall is an assistant professor at the Center for Bioinformatics at Saarland University and affiliated with the Max Planck Institute for Informatics as a senior researcher. He is heading the Algorithms for Computational Genomics group. His research targets algorithmic and statistical challenges arising from present-day genomics technologies. A particular focus is on population-scale sequencing efforts, on structural variation discovery, on algorithms and data structures for pangenomes and on haplotyping in various contexts (from humans to viruses). He has co-organized two workshops on computational pangenomics, one at the Lorentz Center in Leiden, The Netherlands,, which has lead to a comprehensive review paper, and one at ECCB 2016
Tobias received his undergraduate education at Bielefeld University (Germany), where he studied computer science and physics. He obtained his PhD in bioinformatics from TU Dortmund. After that, he moved to Centrum Wiskunde & Informatica (Amsterdam, The Netherlands), the Dutch national institute for mathematics and computer science, as a postdoctoral researcher. During his time as a postdoc, he also was a long-term participant of the semester program on Mathematical and Computational Approaches in High-Throughput Genomics held at the Institute for Pure and Applied Mathematics (IPAM) at University of California Los Angeles (UCLA).

Erik Garrison is a PhD student at the Wellcome Trust Sanger Institute and Cambridge University. His ongoing doctoral research has focused on the development of a software toolkit for practical pangenomics: vg. He has seven years of experience in genomics, where he has worked on the development of sequencing systems, participated in large scale sequencing projects such as the 1000 Genomes Project, and authored popular bioinformatics software such as the freebayes variant caller. Raised in Kentucky, Erik obtained an undergraduate degree in the social sciences from Harvard University. After graduation he worked at Harvard Medical School, One Laptop Per Child, and Boston College before beginning his studies at the Sanger Institute.

Jordan Eizenga is a PhD student in the Computational Genomics Lab at the Unversity of California Santa Cruz. His advisors are David Haussler and Benedict Paten. Jordan was originally trained as a mathematician, but made the switch to bioinformatics in 2015. Now his research focuses on using graph data structures in genomics. He has published on both theoretical and practical aspects of this problem, and he has been heavily involved in the development of the vg project.
- Define the overall learning objectives
- Break down in units (sub-objectives) that can be assigned to instructors
- Identify learning outcomes for each unit.
- For each outcome design an exercise
- For each exercise identify the short lecture needed and the materials
- Compose a schedule, spreading moments of wrap-up, feedback, Q&A, etc.