Working with large trees from the command‐line interface
This guide will show how to use the command-line interface to work with a very large phylogenetic tree. The goal of this tutorial is to perform an analysis similar to the one presented in another example (Displaying BLAST scores), but on a much larger scale. If you have not already done so, please have a look at that example to get an idea of what we are going to do here.
The tree file BchH_ChlH.tre contains an unrooted neighbour-joining tree constructed from 21235 sequences. Of these:
-
20are sequences for the BchH/ChlH gene from various cyanobacteria and anoxygenic phototrophs that were obtained from UniProt. This gene encodes for a subunit of the magnesium chelatase enzyme involved in chlorophyll biosynthesis in photosynthetic bacteria. These sequences were used as queries for a BLAST search. In the tree, these sequences are namedsource_XXX(whereXXXis the original name of the sequence in UniProt, which includes its accession number, source organism and more), for examplesource_tr|Q9F6X9|Q9F6X9_CHLAU Magnesium chelatase OS=Chloroflexus aurantiacus OX=1108 GN=bchH PE=3 SV=1. -
10765are sequences obtained from ablastpsearch on28314bacterial genomes downloaded from RefSeq. The search was performed using the previous20sequences as a query, with a very permissive 10-5 e-value threshold. For each genome in which BLAST reported at least one hit, the best hit (among hits for any of the 20 sequences) was chosen based on the bit score and the corresponding sequence was included in the tree. In the tree, these sequences are namedprot_organism_accession(whereorganismis the scientific name of the organism as reported by RefSeq andaccessionis the RefSeq accession for the genome), for exampleprot_Synechococcus_sp._PCC_7335_GCF_000155595.1. -
10450are sequences obtained from atblastnsearch performed with the same critera (this makes it possible to identify strains where the gene is present in the genome, but has not been annotated in the proteome). In the tree, these sequences are nameddna_organism_accession(whereorganismis the scientific name of the organism as reported by RefSeq andaccessionis the RefSeq accession for the genome), for exampledna_Synechococcus_sp._UTEX_2973_GCF_000817325.1.
Depending on how powerful your computer is, directly opening this tree file in TreeViewer (or in another graphical tree visualisation software) might work, but interacting with it will be a very slow and inefficient task. Instead, we are going to use the command-line interface of TreeViewer to manipulate it.
After you have downloaded the tree file, you should open a command-line window (on Windows, you can do this by opening the Start menu, typing cmd and pressing enter; on macOS you will need to use the Terminal application that you can find in the Utility folder within Applications; on Linux you will need to use your distribution's specific tools) and make sure that the working directory is set to the folder where you have downloaded the file (on all platforms, you should be able to do this by typing cd in the command line - note the space - and then dragging the folder on the command-line window and pressing Enter). Assuming that you have installed TreeViewer using the installer for your platform, you can then start the command-line version of the program by typing TreeViewerCommandLine and pressing enter.
After a few seconds, TreeViewerCommandLine will open, display the version number and wait for your input. Working with TreeViewerCommandline is rather similar to working with the graphical interface of TreeViewer, except that the tree is manipulated by issuing commands in the prompt, rather than by clicking on buttons. As the message issued by the program suggests, you can use the help command to display a list of al the available commands. A command is entered by typing its name (e.g. help) and then pressing Enter. For example, entering the help command should yield an output similar to the following:

To obtain more information about a specific command, you can use the help <command> command. For example, entering help open produces the following output:

You can use the help command to familiarise yourself with the syntax and options of all the other commands that are available in TreeViewerCommandLine (you can even issue help help to get more info about the help command).
As a first step, we can simply draw the tree using the command line interface. To open the tree file, issue the following command:
open BchH_ChlH.tre
Note that tab-completion is available everywhere in the command line. The program will load the file and then ask you if you wish to load the default Transformer and Coordinates modules (i.e. Consensus and Radial, respectively):

Press the Y key to confirm this. The program will then enable these modules and show the default settings:

To enable a new module in TreeViewer, you can use the module enable command. This can also be used for Action modules: for example, issuing the following command (remember you can use tab-completion):
module enable Unrooted tree style
Will have a similar effect as clicking on the Unrooted button in the TreeViewer graphical interface. Instead,
module list enabled
Can be used to show a list of the modules that have currently been enabled:

You can save the plot in PDF or SVG format using the pdf or svg commands, respectively. For example, issuing the command:
pdf BchH_ChlH.pdf
Will create a PDF file containing the tree plot, which should look similar to the following figure:

This figure is not particularly useful, given the huge number of strains in the tree, but at least we were able to produce it without overloading the computer.
Our goal is to produce a figure similar to the one obtained in the Displaying BLAST scores example, i.e. to highlight the BLAST scores on the tree. To do this, the first step is to add the scores to the tree. The BchH_ChlH.data file contains a tab-separated table that includes, for each strain, the % identity to the query sequence, the alignment length, the e-value, and the bit score. The file should look like the following, when opened in a text editor:
Genome PercentIdentity AlignmentLength EValue BitScore
dna__Massilia_aquatica__Holochova_et_al._2020_GCF_011682045.1 29.078 1269 2.43E-145 489
dna__Massilia_aquatica__Lu_et_al._2020_GCF_009857595.1 31.077 724 4.82E-77 279
dna__Nostoc_azollae__0708_GCF_000196515.1 87.735 1329 0 2439
dna_Acaryochloris_marina_MBIC11017_GCF_000018105.1 80.15 1330 0 2219
dna_Acaryochloris_sp._CCMEE_5410_GCF_000238775.1 80.226 1330 0 2218
...
The file can be added as an Attachment from the command line by issuing the following command:
attachment add BchH_ChlH.data
The program will then ask for a name for the attachment (e.g. BchH_ChlH) and then will ask two more questions, to which you should reply Yes (i.e. press Y). Just as we did in the previous example, to actually associate the various values contained in the data file to the tree, we need to use the Parse node states module. To enable this module, issue the command:
module enable Parse node states
This will enable the new module and show its current settings:

The settings can be changed using the option command. Enter:
option select Data file
To select the Data file option, then enter:
option set BchH_ChlH
To set the value of this parameter to the attachment that we have just added to the tree. Now, enter:
option select Use first row as header
To select the check box (remember you can use tab completion) and enten enter:
option set true
To check the check box. Now, the module has been set up to associate the scores to the tree. You can display the current values of the parameters for the selected module by using the following command:
option list

After making changes to the options for a further transformation module, you need to issue the update command in order to apply the changes; this is like clicking on the Apply button in the graphical interface. If you do not invoke this command, the next time you try to enable a module, the program will complain that there are pending changes. Therefore, enter the command:
update
This may take a couple of seconds. Now, as was the case in the other example, the query sequences have not been assigned a score, because they do not appear in the data file (as they are not the result of a BLAST search). Thus, before going further, we need to assign a fictitious score to them. To do this, we can use the Replace attribute module. To enable this module, issue the following command:
module enable Replace attribute
This will add the module and print its options. This module has two options with the same name (Attribute), i.e. the search Attribute and the replacement Attribute; this means that we cannot select them using their name, and we must instead resort to the option number. To set up the options for this module, issue the following commands:
option select #1
option set Name
option select #3
option set source_
option select #8
option set BitScore
option select #9
option set Number
option select #10
option set 3000
update
This will set up the module so that it matches taxa with source_ in their Name (i.e. the query sequences) and adds to them a numeric attribute called BitScore with value 3000. You can use option list to show the new values for all the options:
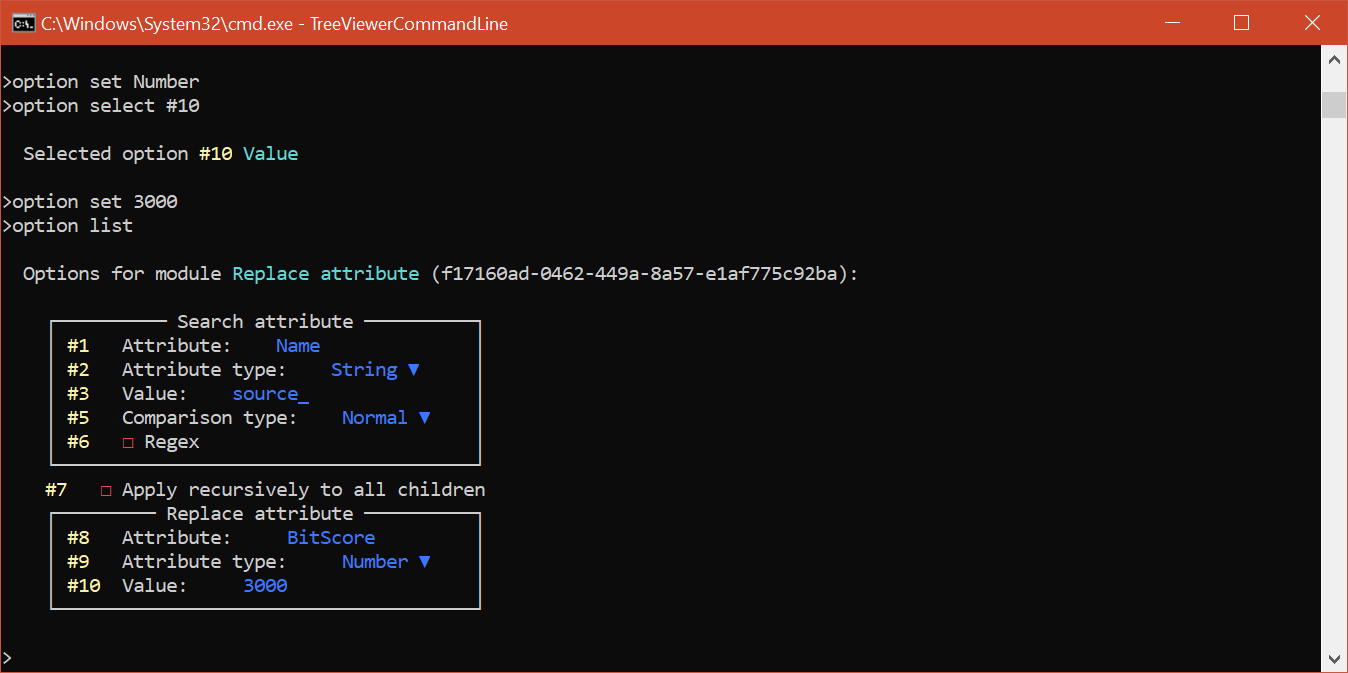
Again, as was the case in the other example, we need to use the Propagate attribute module to propagate the bit scores to the internal branches. To enable this module and set up its options, you can issue the following commands:
module enable Propagate attribute
option select Attribute
option set BitScore
update
We are now ready to draw the tree highlighting the branch scores. In the graphical interface, we would click on the Branch scores button; here, instead, we can enable the Branch score style module from the command line:
module enable Branch score style
Once you issue this command, the program will ask you a number of questions that is equivalent to the choices that would be presented to you in the window that opens when you click on the Branch scores button in the interface. For each question, there is a default value that is highlighted: if you wish to choose it, you just have to press Enter without entering any text.
The first question is the attribute that you would like to use for the branch scores. The default choice should already be the BitScore attribute, so you can just press Enter here. Then, you need to enter the score range (by entering first the minimum and then the maximum score). You should enter a minimum of 0 and a maximum of 1000. You can also use the default value for all the remaining questions.
Now, make sure that the PDF plot that was produced earlier is not open in another program, and issue the command:
pdf
The pdf command, when issued without an argument, saves the plot to the same file as the last time it was invoked. In this case, it should overwrite the BchH_ChlH.pdf file that you created earlier. The new figure should look similar to the following:
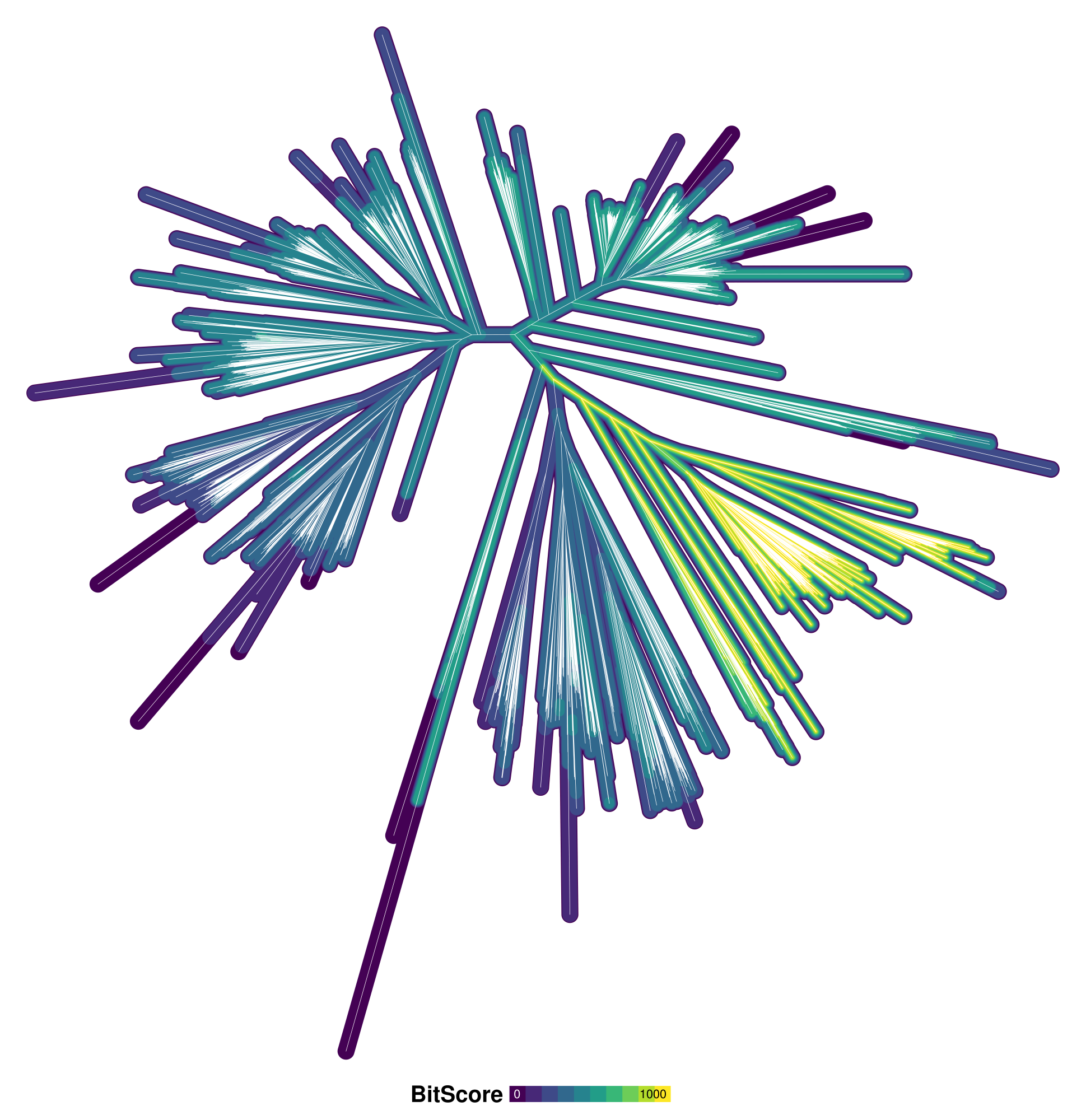
If you are on Windows, a program such as SumatraPDF will let you view the PDF plot without "locking" it, i.e. the file can still be overwritten, and the program will automatically refresh whenever it is updated. On macOS, you can obtain a similar result using the included Preview app. On Linux, you can use something like Evince.
From this plot it should be clear which part of the tree contains the "true orthologs"; however, we should still highlight the query sequences, to make sure that they are in the right place. To do this, we are going to use another instance of the Replace attribute module, which will assign a numeric attribute called Query with value 150 to the query sequences. This can be achieved by issuing the following commands:
module enable Replace attribute
option select #1
option set Name
option select #3
option set source_
option select #8
option set Query
option select #9
option set Number
option select #10
option set 150
update
This is similar to the Replace attribute module that we used earlier to set the bit score for the query sequences. If you issue option list, you can check that all the options have been set to the correct value:

We can now add a Node shapes module that will draw a star at the query sequences. As we did in the other example, we will set the default shape Size to 0, and allow it to be overridden by the Query attribute, so that the node shapes only appear at the query sequences. We will also disable the Auto fill colour by node option, so that all the stars have the same colour, and give them a white contour. To set this up, issue the following commands:
module enable Node shapes
option select Size
option set 0
option set attribute number Query
option select Auto fill colour by node
option set false
option select Stroke thickness
option set 10
option select Stroke colour
option set #FFFFFF
Once again, by issuing option list you can check the parameter values:

You can now update the plot again:
pdf
The new module should have caused some blue stars to appear on top of the branches representing the query sequences:

We can now confidently say that the "true orthologs" are found in the green-yellow area of the tree to the right. However, we still need a way to select the tips of the tree that are in this area.
We cannot open this tree directly in TreeViewer, because the program would try to draw the tree together with the branch labels, and that would take a very long time. The trick that we are going to use is to open the tree again in TreeViewerCommandLine, and set it up so that only the branches are drawn; we can then export it in a format that preserves the information about active modules, and open this new tree file with TreeViewer: the program, seeing that the file mandates only for branches to be drawn, will not try to draw the tip labels, and this will improve the performance sensibly.
To do this, open another command-line session with TreeViewerCommandLine (keep the other one open, we will need it later) and again open the tree file:
open BchH_ChlH.tre
Then, press Y to confirm that you want to load the default modules and enable the Unrooted tree style Action module:
module enable Unrooted tree style
update
Now, we can remove the labels from the plot by disabling the Labels module:
module disable Labels
Before exporting the tree file, we ought to highlight the query sequences here as well, since this will make it easier to identify the region of the tree corresponding to the true orthologs. You can use the commands from the previous steps to add a Replace attribute Further transformation and a Node shapes Plot action to do this:
module enable Replace attribute
option select #1
option set Name
option select #3
option set source_
option select #8
option set Query
option select #9
option set Number
option select #10
option set 150
update
module enable Node shapes
option select Size
option set 0
option set attribute number Query
option select Auto fill colour by node
option set false
We can now export the tree file in a format that preserves the module information, e.g. in Binary tree format. To do this, you can use the binary command:
binary modules loaded BchH_ChlH_simple.tbi
Press Y when you are asked whether you want to sign the file. This command will export the loaded tree along with the Transformer, Further transformation, Coordinates and Plot action modules to a file in Binary tree format called BchH_ChlH_simple.tbi. The BchH_ChlH_simple.tbi can now be opened directly in the graphical version of TreeViewer, and hopefully should not take too long to load. You can close the second TreeViewerCommandLine interface (i.e. the one we just used to create the simple tree file) by typing:
exit
In the TreeViewer graphical interface (once the tree loads and is drawn), click on the Lasso selection button under the Actions to enable the lasso selection, then draw a shape around the part of the tree that contains the "true orthologs":

The window that opens should tell you that 2960 tips and 5919 nodes have been selected; make sure that the Copy attribute at option is set to Tips and that the attribute to copy is set to Name and click on OK. This will copy the names of the 2960 selected tips to the system clipboard; you can now open a simple text editor and paste them. Save the resulting text file in the same folder as the tree, calling it e.g. orthologs.txt.
You can now close the graphical version of TreeViewer and go back to the command-line version that we were using to produce the actual plot. Here, we want to load the new file containing the names of the orthologs as an Attachment, and then use it to add an attribute to the corresponding tips of the tree, so that we can highlight them as well.
To add the file as an attachment, you can issue the following command:
attachment add orthologs.txt
Again, you will have to enter a name for the attachment (e.g. orthologs) and press Y twice to answer the questions. Before doing anything else, we need to update the state of the plot:
update
We now need to add the Add attribute module to add an attribute to the "true orthologs". However, we have a problem: there are two modules with the same name "Add attribute". Indeed, if you try to run the following command:
module enable Add attribute
You will receive a message saying that the module selection is ambiguous, and suggesting to use the module ID instead of the name, to univocally specify the module you want to enable. We can get a list of the available Further transformation modules by running the following command:
module list available Further transformation

Here, we can see that there are two Add attribute modules, one with Id afb64d72-971d-4780-8dbb-a7d9248da30b and one with Id f71a5e60-5e40-4a5e-9795-e5259fb283ab. To understand which one of these is the module we need, we can use the module help command:
module help afb64d72-971d-4780-8dbb-a7d9248da30b
module help f71a5e60-5e40-4a5e-9795-e5259fb283ab

This command prints the brief description of a module. This should make it clear that the module we need is the one with Id f71a5e60-5e40-4a5e-9795-e5259fb283ab. Therefore, we can enable this module (remember that you can just enter the first characters of the Id and then use tab-completion to let the program figure out the rest):
module enable f71a5e60-5e40-4a5e-9795-e5259fb283ab
Now, we can set the parameters for this module:
option select Taxon list
option set orthologs
option select Attribute
option set Ortholog
option select Attribute type
option set Number
option select New value
option set 50
update
These options will associate a new attribute called Ortholog to the taxa whose Name is in the attachment. As usual, you can issue option list to check that the correct values have been entered:

Now, we need to add another Node shapes module to highlight the orthologs:
module enable Node shapes
As before, we are going to set the default Size to 0 and associate it with the new Ortholog attribute; we are also going to disable the Auto fill colour by node and give the shapes a white contour:
option select Size
option set 0
option set attribute number Ortholog
option select Auto fill colour by node
option set false
option select Stroke thickness
option set 3
option select Stroke colour
option set #FFFFFF
Now, if you issue option list, you will notice that having disabled the Auto fill colour by node option caused a new option Fill colour to appear:

To make sure that the query sequences and the true orthologs have different colours, we can change the value of this option:
option select Fill colour
option set #D55E00
This will set the fill colour to an orange hue. You can now update the plot:
pdf

The query sequences and the true orthologs are now both highlighted on the tree; however, since the symbols highlighting the orthologs are many more (and much smaller) than the ones for the query sequences, it would be better if they were below the query sequence markers, instead of above them. We can achieve this in a similar way as we would achieve it if we were using the graphical version of TreeViewer, i.e. by "moving" up the second Node shapes module. First of all, since there are two Node shapes modules in the plot, we need to get a list of all the modules that are currently enabled:
module list enabled
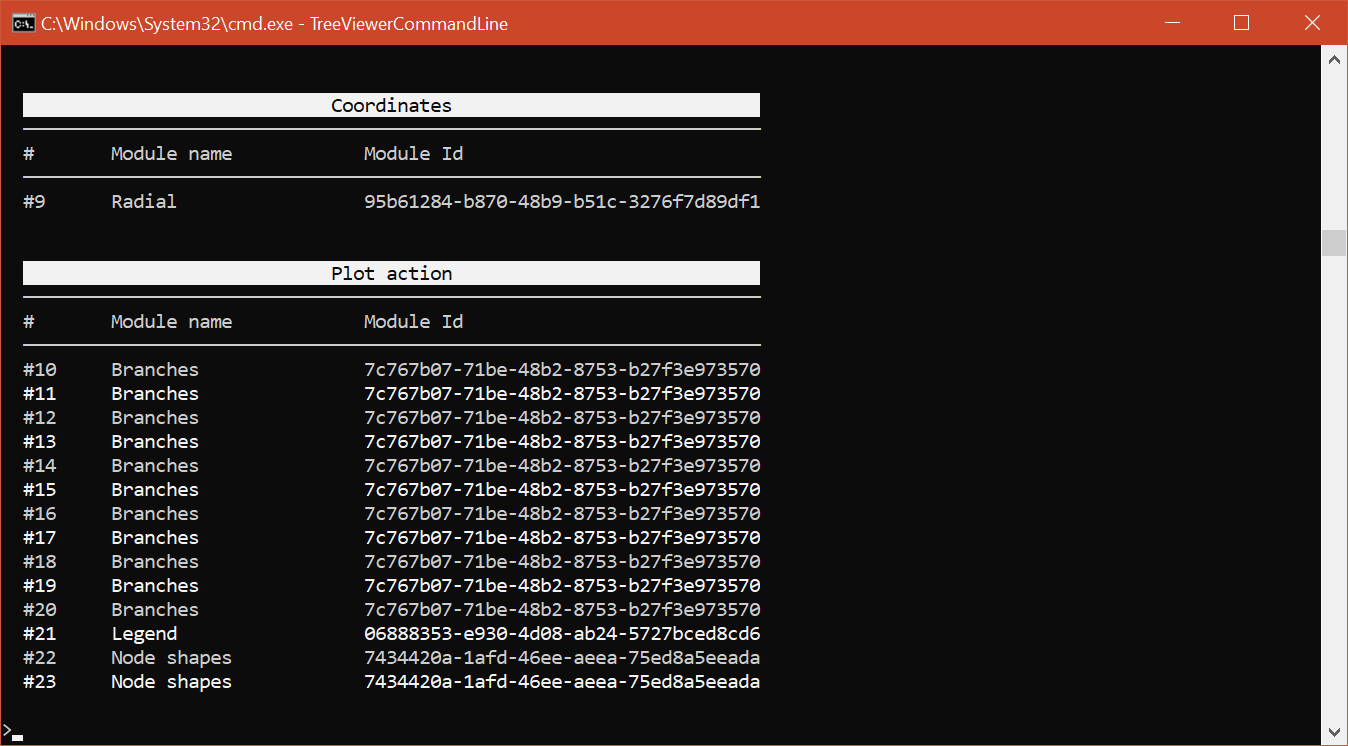
From here we can clearly see that we need to move up module #23 (or to move down module #22). This can be done using the module move command:
module move up #23
We can now update the plot:
pdf

As usual, the last thing that remains is to update the legend. To do this, first of all select the Legend module that was added when we used the Branch score style action module:
module select Legend
Then, you can list the options for this module:
option list

First of all, we need to change the Markdown source of the legend:
option select Markdown source
option set source
This will open a (command-line) text-editor window that you can use to enter the Markdown source. Delete all the text that is currently present and replace it with the following:
# **Legend**
### Bit score 
###  Orthologs
###  Query sequence

This is exactly the same code that we used in the Displaying BLAST scores example. When you have finished, press CTRL+X (on all platforms) to save the file, press Y to confirm, and then press Enter to overwrite the existing file. One last thing that we can do before plotting the tree again is to change the position of the legend. Right now, it sits at below the tree; however, there is quite a bit of space available in the bottom right corner, so it does not make sense to have the legend occupy more space than necessary. To position the legend in the bottom-right corner, use the following commands:
option select Anchor
option set Bottom-right
option select Alignment
option set Bottom-right
option select Position
option set 0, 0
These will align the bottom-right corner of the legend with the bottom-right corner of the plot and reset the position. You can now plot the tree again:
pdf
The final plot should look similar to the following figure:

You can now save the tree file using the binary command:
binary modules loaded BchH_ChlH.tbi
This command will save the tree, including all the modules that have been enabled, as well as all the attachments (answer Y to both questions). You can also download the BchH_ChlH.tbi tree file, which contains the tree along with all the modules. You probably do not want to open this file with the graphical version of TreeViewer, as they are likely too heavy to be handled in this way (maybe unless you are reading this a few years from when this was written...); instead, only use them with TreeViewerCommandLine.
-
As noted before, if you do not want to continuously open and close the PDF file, you should use a PDF viewer that does not "lock" it: if you are on Windows, you can use SumatraPDF; on macOS, you can use the Preview app; on Linux, you can use something like Evince. The Adobe PDF viewer instead will not be appropriate for this use, because it does lock the file and prevents other programs from updating it.
-
You can also integrate TreeViewerCommandLine in a pipeline of command-line programs: you just need to create a text file (called e.g.
plot.txt) containing the commands you want to issue to the program, and run it piping the contents of the text file to the standard input of TreeViewerCommandLine. For example, to create the plot we have just produced, you can use a text file with the following commands:open BchH_ChlH.tre y attachment add BchH_ChlH.data BchH_ChlH y y update module enable Parse node states option select Data file option set BchH_ChlH option select Use first row as header option set true update module enable Replace attribute option select #1 option set Name option select #3 option set source_ option select #8 option set BitScore option select #9 option set Number option select #10 option set 3000 update module enable Propagate attribute option select Attribute option set BitScore update module enable Branch score style BitScore 0 1000 10 Viridis update module enable Replace attribute option select #1 option set Name option select #3 option set source_ option select #8 option set Query option select #9 option set Number option select #10 option set 150 update attachment add orthologs.txt orthologs y y update module enable f71a5e60-5e40-4a5e-9795-e5259fb283ab option select Taxon list option set orthologs option select Attribute option set Ortholog option select Attribute type option set Number option select New value option set 50 update module enable Node shapes option select Size option set 0 option set attribute number Ortholog option select Auto fill colour by node option set false option select Stroke thickness option set 3 option select Stroke colour option set #FFFFFF option select Fill colour option set #D55E00 module enable Node shapes option select Size option set 0 option set attribute number Query option select Auto fill colour by node option set false option select Stroke thickness option set 10 option select Stroke colour option set #FFFFFF module select Legend option select Markdown source option set source legend.md option select Anchor option set Bottom-right option select Alignment option set Bottom-right option select Position option set 0, 0 binary modules loaded BchH_ChlH.tbi y y pdf BchH_ChlH.pdfYou can also download
plot.txt. Make sure that you have a single folder containing:- The
plot.txtfile with the commands - The
BchH_ChlH.tretree file - The
BchH_ChlH.datadata file - The list of orthologs
orthologs.txt - A file called
legend.mdthat contains the Markdown code that will be used to draw the legend (you can copy and paste the text from above, or you can downloadlegend.md)
Now, you can run TreeViewerCommandLine and tell it to read the commands from the input file by executing from your command-line interface:
TreeViewerCommandLine < plot.txtTreeViewerCommandLine will basically repeat all the steps that were involved in this tutorial and produce the
BchH_ChlH.pdfPDF plot and theBchH_ChlH.tbiBinary tree file.This approach is powerful because, naturally, you could generate the
plot.txtfile using other steps in your pipeline (or, you could have a "skeleton" file in which you replace some commands as necessary). If you connect the standard output of another process to the standard input of TreeViewerCommandLine, you could even have another program communicate "directly" with TreeViewerCommandLine. - The