This repository contains the implementation code for our preprint Aggregating Residue-Level Protein Language Model Embeddings with Optimal Transport, which showcases the benefits of sliced-Wasserstein embedding to summarize token-level representations produced by pre-trained protein language models (PLMs), including ESM-2 and ProGen2.
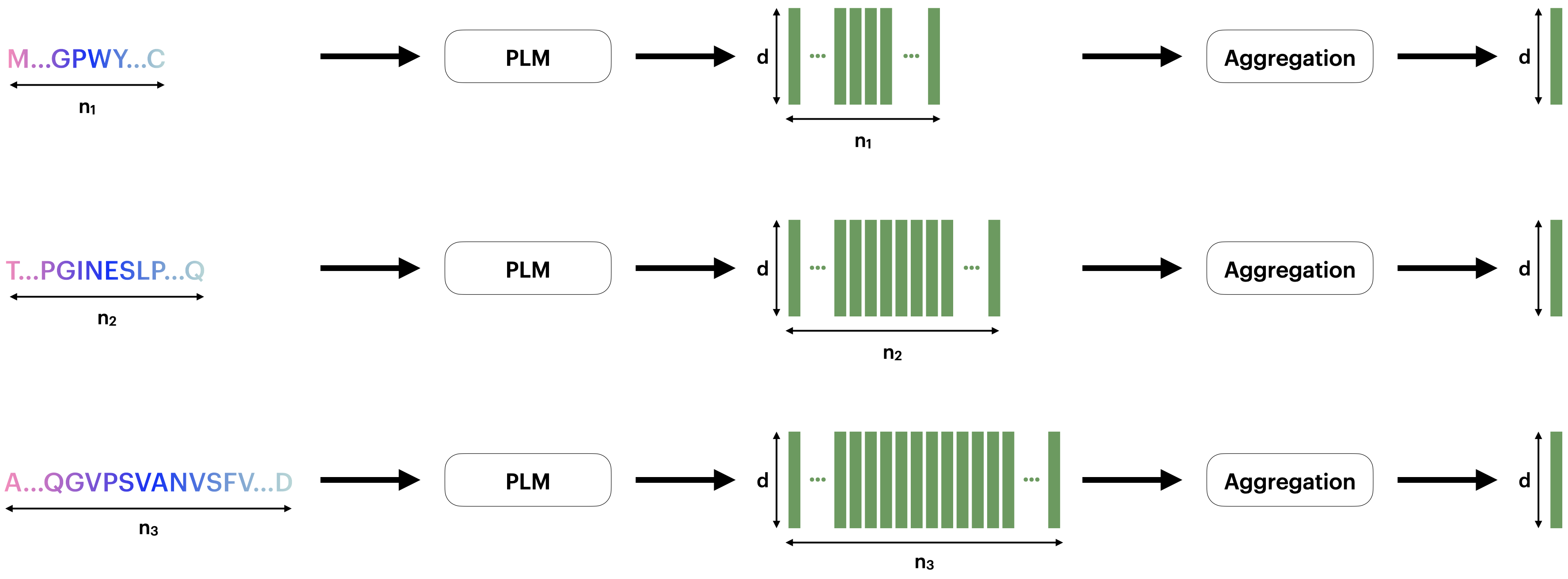
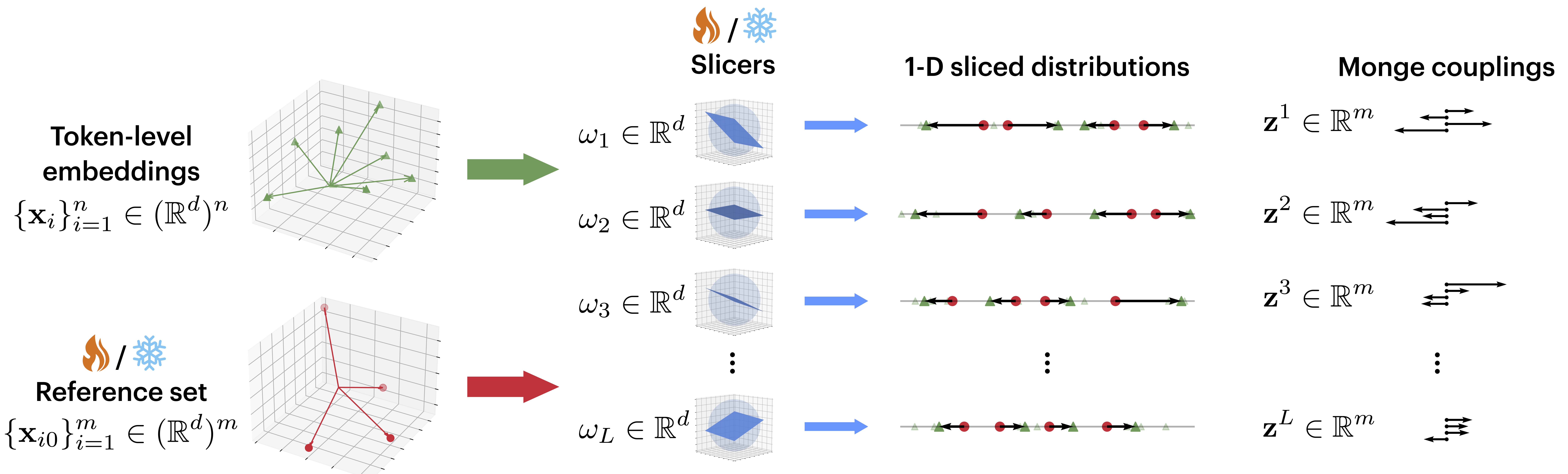
Protein language models (PLMs) have emerged as powerful approaches for mapping protein sequences into embeddings suitable for various applications. As protein representation schemes, PLMs generate per-token (i.e., per-residue) representations, resulting in variable-sized outputs based on protein length. This variability poses a challenge for protein-level prediction tasks that require uniform-sized embeddings for consistent analysis across different proteins. Previous work has typically used average pooling to summarize token-level PLM outputs, but it is unclear whether this method effectively prioritizes the relevant information across token-level representations. We introduce a novel method utilizing optimal transport to convert variable-length PLM outputs into fixed-length representations. We conceptualize per-token PLM outputs as samples from a probabilistic distribution and employ sliced-Wasserstein distances to map these samples against a reference set, creating a Euclidean embedding in the output space. The resulting embedding is agnostic to the length of the input and represents the entire protein. We demonstrate the superiority of our method over average pooling for several downstream prediction tasks, particularly with constrained PLM sizes, enabling smaller-scale PLMs to match or exceed the performance of average-pooled larger-scale PLMs. Our aggregation scheme is especially effective for longer protein sequences by capturing essential information that might be lost through average pooling.
Create a new virtual Conda environment, called plm_swe, with the required libraries using the following commands:
conda create -n plm_swe python=3.9
conda activate plm_swe
pip install -r requirements.txt
The download_data.py script can be used to download the datasets for the experiments into a new folder called datasets by running
python download_data.py --to datasets --benchmarks davis bindingdb scl ec ppi_gold
The download_progen2.sh script can be used to download the pre-trained ProGen2 PLMs into the progen2-checkpoints folder by running
bash download_progen2.sh
The following commands can be used to run the numerical experiments in the paper. The number of points in the reference set and the pre-trained PLM backbone can be adjusted via command-line parameters, as well as the configuration files under config.
python run_dti.py --run-id dti_davis_swepooling_100refpoints_freezeTrue_esm2_8m --config config/dti_davis_esm2.yaml --pooling swe --num-ref-points 100 --freeze-swe True --target-model-type esm2_t6_8M_UR50D
python run_dti.py --run-id dti_davis_swepooling_100refpoints_freezeTrue_progen2_small --config config/dti_davis_progen2.yaml --pooling swe --num-ref-points 100 --freeze-swe True --target-model-type progen2-small
python run_dti.py --run-id dti_bindingdb_swepooling_100refpoints_freezeFalse_esm2_8m --config config/dti_bindingdb_esm2.yaml --pooling swe --num-ref-points 100 --freeze-swe False --target-model-type esm2_t6_8M_UR50D
python run_dti.py --run-id dti_bindingdb_swepooling_100refpoints_freezeFalse_progen2_small --config config/dti_bindingdb_progen2.yaml --pooling swe --num-ref-points 100 --freeze-swe False --target-model-type progen2-small
python run_dti.py --run-id dti_tdc_dg_swepooling_100refpoints_freezeTrue_esm2_8m --config config/dti_tdc_dg_esm2.yaml --pooling swe --num-ref-points 100 --freeze-swe True --target-model-type esm2_t6_8M_UR50D
python run_scl.py --run-id scl_swepooling_100refpoints_freezeFalse_esm2_8m --config config/scl_esm2.yaml --pooling swe --num-ref-points 100 --freeze-swe False --target-model-type esm2_t6_8M_UR50D
python run_ec.py -c config/ec_esm2.yaml --modeldir ec/model --pooling swe --num_swe_ref_points 100 --freeze_swe False --model ESM-2-8M --seed 12345
python run_ppi.py --run-id ppi_gold_swepooling_128refpoints_128slices_esm2_8m --config config/ppi_gold_esm2.yaml --num-ref-points 128 --num-slices 128 --target-model-type esm2_t6_8M_UR50D
This repository is built upon the following GitHub repositories:
If you make use of this repository, please cite our preprint using the following BibTeX format:
@article{naderializadeh2024_plm_swe,
title={Aggregating Residue-Level Protein Language Model Embeddings with Optimal Transport},
author={NaderiAlizadeh, Navid and Singh, Rohit},
journal={bioRxiv},
year={2024},
publisher={Cold Spring Harbor Laboratory}
}