RnaSeq
- Process Flow
- Description
- What is RNA-seq?
- Overview
- How to analyze RNA-seq data?
- Output Files and Formats
- Frequently Asked Questions
Processing Profile: 2762841

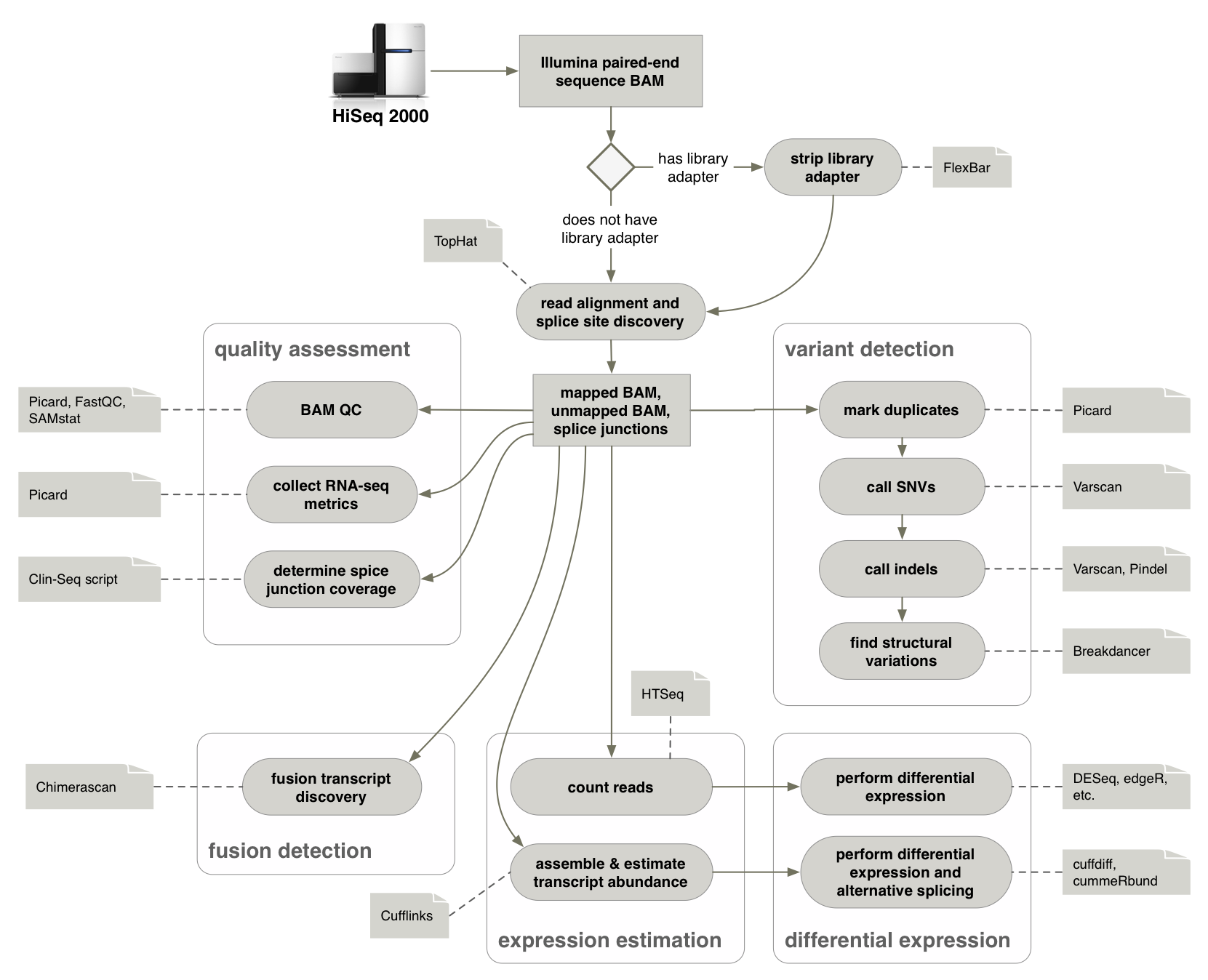
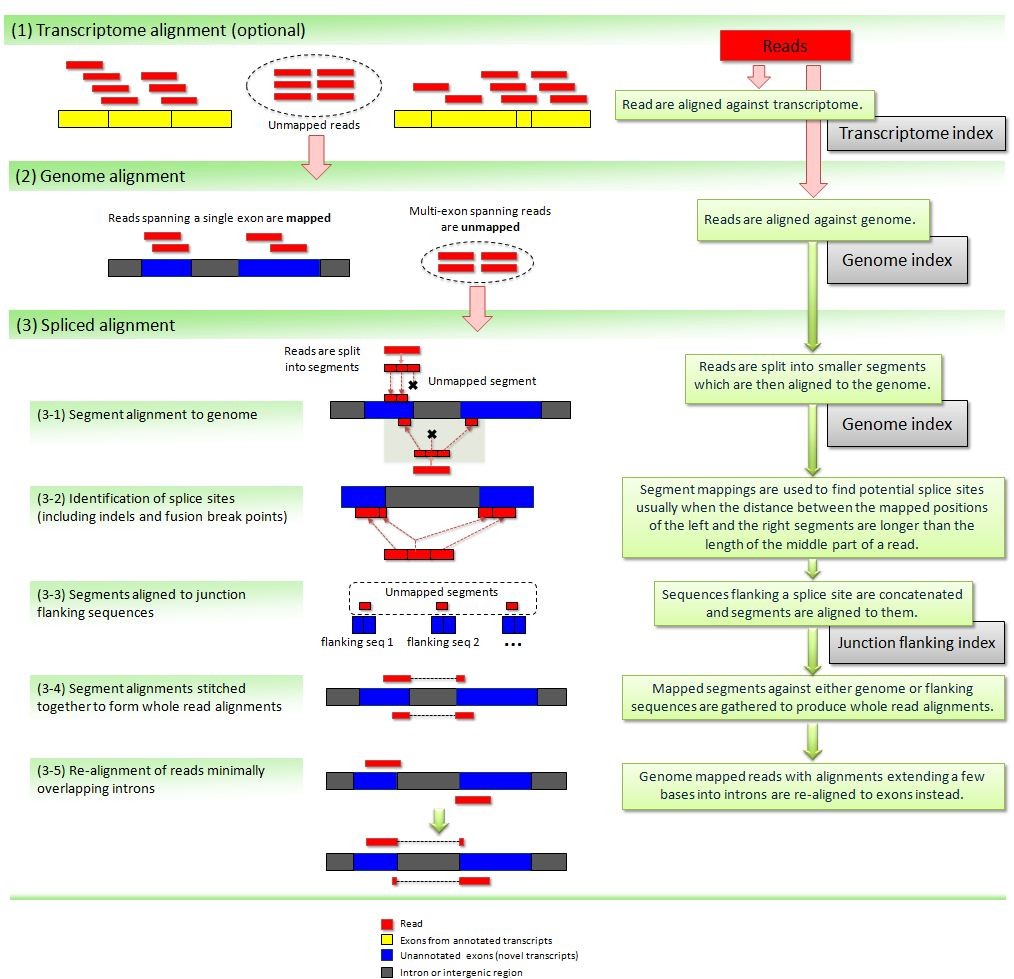
Each lane or sub-lane of data is aligned to GRCh37-lite with Ensembl 74 annotation using TopHat 1,2,3 v2.0.4 which performs mappings using Bowtie2 v2.0.0 after "bisecting" each read for optimal cross-exon placement. The only variation from default command line parameters is setting the number of cores and bowtie version (-p 4 --bowtie-version=2.0.0). Downstream BAM file conversion and manipulation performed with Picard4 v1.52.
Picard CollectRnaSeqMetrics is performed using the Ensembl v74 annotation and the GRCh37-lite reference to assess the amount of the mapped reads that align to the genomic components (ex. intergenic, intronic, coding, utr, and ribosomal or mitochondrial).
Resultant TopHat mappings are processed through Cufflinks 4,5,6,7 v2.0.2 which assembles aligned RNA-seq reads into transcripts and estimates their abundances. Cufflinks is run in three modes using the annotation in various ways to identify abundance of a de novo transcript assembly(default), known transcripts only(-G), and a reference-guided hybrid mode (-g). Cufflinks is run using Ensembl v74 annotation to help drive transcript identification in the both the known transcripts and reference-guided modes. For all three modes abundance estimates are made using FPKM (Fragments Per Kilobase of exon model per Million mapped fragments) values.
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics doi:10.1093/bioinformatics/btp120
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology 10:R25.
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. . Genome Biology 2011, 14:R36
- http://picard.sourceforge.net.
- Trapnell C, Williams BA, Pertea G, Mortazavi AM, Kwan G, van Baren MJ, Salzberg SL, Wold B, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation Nature Biotechnology doi:10.1038/nbt.1621
- Roberts A, Trapnell C, Donaghey J, Rinn JL, Pachter L. Improving RNA-Seq expression estimates by correcting for fragment bias Genome Biology doi:10.1186/gb-2011-12-3-r22
- Roberts A, Pimentel H, Trapnell C, Pachter L. Identification of novel transcripts in annotated genomes using RNA-Seq Bioinformatics doi:10.1093/bioinformatics/btr355

- Determine Model Inputs a) Instrument Data (The first library name starts with H_KH-540077-Tumor-cDNA-8):
$ genome instrument-data list solexa --filter library_name=Pooled_RNA_2891007020-mRNA2-cDNA-1-lig1-lib1
ID FLOW_CELL_ID LANE INDEX_SEQUENCE SAMPLE_NAME LIBRARY_NAME READ_LENGTH IS_PAIRED_END CLUSTERS MEDIAN_INSERT_SIZE SD_ABOVE_INSERT_SIZE TARGET_REGION_SET_NAME
-- ------------ ---- -------------- ----------- ------------ ----------- ------------- -------- ------------------ -------------------- ----------------------
2891354555 C1TD1ACXX 8 ACAGTG H_NJ-HCC1395-HCC1395_RNA Pooled_RNA_2891007020-mRNA2-cDNA-1-lig1-lib1 100 1 156248832 247 1421 <NULL>
b) Process Profile (current defaults/only option) is : 2762841 (October 2012 Default Ovation V2 RNA-seq)
c) Reference Sequence: 106942997
d) Annotation: 124434505
- Create a Genome Model Group to help organize models:
$ genome model-group create $USER-rnaseq-tutorial-test
Created model group:
ID: 8a6d9e68ceed4c96af3cc36d6b321cd6, NAME: gmsuser-rnaseq-tutorial-test
3. Define a Genome Model:
$ genome model define rna-seq --groups=8a6d9e68ceed4c96af3cc36d6b321cd6 --reference-sequence-build=106942997 --annotation-build=124434505 --instrument-data=2891354555 --processing-profile=2762841
'instrument_data', 'annotation_build', 'groups', 'reference_sequence_build', and 'processing_profile' may require verification...
Resolving parameter 'instrument_data' from command argument '2891354555'... found 1
Resolving parameter 'annotation_build' from command argument '124434505'... found 1
Resolving parameter 'groups' from command argument '8a6d9e68ceed4c96af3cc36d6b321cd6'... found 1
Resolving parameter 'reference_sequence_build' from command argument '106942997'... found 1
Resolving parameter 'processing_profile' from command argument '2762841'... found 1
Created model:
id: 4db14ea02335464dab7499b83b4a50d4
name: H_NJ-HCC1395-HCC1395_RNA.rna_seq
subject: H_NJ-HCC1395-HCC1395_RNA (TST1 tumor 2889953342)
processing_profile: Default Ovation V2 RNA-seq (2762841)
4. Start a Build of the Genome Model
$ genome model build start 8a6d9e68ceed4c96af3cc36d6b321cd6
6. Interrogate Build Results (see next section)
Sometimes you may have RNA-seq data that you want to analyze with a non-Ensembl set of transcripts. If you have a gtf file, you should be able to do this by creating an rna-seq only imported annotation build.
The resulting build can be used as the --annotation-build input to the RNA-seq model.
This is not a comprehensive overview of RNA-seq analysis, but rather a primer on the RNA-seq pipeline:
| Name | Type | Description |
|---|---|---|
| build.xml | Text File | Workflow XML file used by the GMS to run the analysis on the TGI cluster. |
| alignments | Symbolic Link | GMS Alignment API software result. |
| alignment_stats | Symbolic Link | An in-house tool is used to generate alignment metrics about the BAM file |
| metrics | Symbolic Link | Picard CollectRnaSeqMetrics software result |
| junctions | Symbolic Link | An in-house tool used to summarize splice junction coverage. |
| bam-qc | Symbolic Link | Software result containing general quality control data including: Picard, Samtools, SAMStat, FastQC, and in-house tools. |
| logs | Directory | The standard error and standard out of all applications and workflow commands used to generated the build. |
| de_novo | Symbolic Link | The 'de novo' mode Cufflinks software result. |
| reference_only | Symbolic Link | The 'reference only' mode Cufflinks software result. |
| expression | Symbolic Link | The 'reference only' mode Cufflinks software result. Retained for backward compatibility. |
| reference_guided | Symbolic Link | The 'reference guided' mode Cufflinks software result. |
| results | Directory | A base-level results directory. Created to provide consistency with newer build data directories. |
| fusions | Directory | A base-level directory for fusion detection results. |
| reports | Directory | The auto-generated pipeline reports created by the GMS. Typically these are only relevant to a build's success or failure. |
The files contained in the alignments/ directory are the product of the alignment of all sequence reads to a reference genome. Each instrument data is aligned independently and each result exists on the filesystem. The individual alignment results are not linked to the build data directory, but are currently retained. Please see the aligner documentation for the contents of the individual alignment results.
| Name | Type | Description |
|---|---|---|
| *.bam | Binary file | The merged, position sorted, and optionally marked duplicate BAM file of alignments. |
| *.bam.bai | Binary File | An index of the BAM file. |
| *.bam.md5 | Text File | The md5 checksum of the BAM file. |
| *.bam.flagstat | Text File | Samtools flagstat output for the BAM file. See the Alignment Stats section for notes. |
| *.log | Text File | (optional) Per-library log files from Picard MarkDuplicates. |
| *.metrics | Text File | (optional) Per-library metric files from Picard MarkDuplicates. |
Transcriptome alignment is not quite as simple as genome alignment. RNAseq reads are typically allowed to align to multiple locations. Therefore, the SAMTools flagstat output is not useful. It only accounts for the number of alignments in a BAM file rather than the number of reads. A tool was developed at The Genome Institute to uniquely count the number of reads and metrics about the alignment of those reads. The tool is a GMS command with a package name of Genome::Model::Tools::BioSamtools::Tophat2AlignmentStats.
| Name | Type | Description |
|---|---|---|
| alignment_stats.txt | Text File | A tab-separated value file of alignment metrics per chromosome. A footer exists with summary metrics prefixed by '##'. |
The tab-delimited columns described here are written per chromosome for the reference:
| Column | Header | Description |
|---|---|---|
| 1 | chr | The reference genome chromosome for which each metric is reported. |
| 2 | top | Reads with one, unique alignment to the genome. |
| 3 | top-spliced | Spliced, aligned reads with one, unique alignment to the genome. |
| 4 | pct_top_spliced | A percentage of the top reads with spliced alignment. |
| 5 | multi_reads | Reads with multiple alignments to the genome. |
| 6 | pct_multi_reads | A percentage of the total reads that map to multiple locations. |
| 7 | multi_spliced_reads | Spliced, aligned reads with more than one alignment to the genome. |
| 8 | pct_multi_spliced_reads | A percentage of the multiple alignment reads that are spliced alignment. |
| 9 | multi_hits | The total number of alignments that multiple mapped reads consume. |
the '##' prefixed summary metrics are for the entire reference genome:
| Name | Description |
|---|---|
| Total Reads | Total number of reads in the BAM file. |
| Unmapped Reads | Number of unmapped reads in the BAM file. |
| Unique Alignments | Alignments contained in the BAM file that are uniquely mapped. |
| Multiple Hit Reads | Reads in the BAM file that have multiple alignments. |
| Multiple Hit Sum | The sum of the number of alignments consumed by multiply aligned reads. |
| Total Reads Mapped | Number of reads that are mapped, either uniquely or with multiple alignments. |
All of the files in the metrics/ directory are a product of Picard CollectRnaSeqMetrics. For a complete definition of each metrics see the Picard Metric Definitions page.
| Name | Type | Description |
|---|---|---|
| PicardRnaSeqChart.pdf | PDF File | Normalized RNA−Seq Coverage vs. Normalized Transcript Position for the top 1,000 expressed genes. |
| PicardRnaSeqMetrics.png | Image File | Three dimensional pie chart of the proportion of aligned data that belongs to coding, UTR, intronic, intergenic and mitochondrial/ribosomal annotation. |
| PicardRnaSeqMetrics.txt | Text File | A text file of RNA-Seq metrics including basic alignment summary, proportion of alignments to annotation, transcript bias and strand specificity. |
| Picard_mRNA.refFlat | Text File | refFlat annotation file used by the Picard tool to collect RNA-Seq metrics. Converted from GTF using gtfToGenePred and then an in-house tool to print the first 9 columns as refFlat. |
| Picard_ribo.intervals | Text File | The IntervalList annotation file of mitochondrial and ribosomal annotation used by the Picard tool to collect RNA-Seq metrics. |
| Name | Type | Description |
|---|---|---|
| AlignmentResult_*_junctions.bed | Text File | |
| *.ExonContent.bed | Text File | |
| *.Exons.bed | Text File | |
| *.Genes.bed | Text File | |
| *.Junction.GeneExpression.top1percent.tsv | Text File | |
| *.Junction.GeneExpression.tsv | Text File | |
| *.Junction.TranscriptExpression.top1percent.tsv | Text File | |
| *.Junction.TranscriptExpression.tsv | Text File | |
| *.junctions.bed | Text File | |
| *.junctions.tsv | Text File | |
| SpliceJunctionSummary.R.stderr | Text File | |
| SpliceJunctionSummary.R.stdout | Text File | |
| junctions.bed | Text File | |
| observed.junctions.anno.*.tsv | Text File | |
| observed.junctions.bed | Text File | |
| summary | Directory |
| Name | Type | Description |
|---|---|---|
| ObservedJunctions_SpliceSiteUsage_Pie.pdf | PDF File | |
| KnownJunctions_SpliceSiteUsage_Pie.pdf | PDF File | |
| ObservedJunctions_SpliceSiteAnchorTypes_Pie.pdf | PDF File | |
| ObservedJunctions_ReadCounts_Log2_Hist.pdf | PDF File | |
| GeneJunctionReadCounts_Log2_Hist.pdf | PDF File | |
| TranscriptJunctionReadCounts_Log2_Hist.pdf | PDF File | |
| KnownJunctionCounts_Genes_Log2_Hist.pdf | PDF File | |
| KnownJunctionCounts_Transcripts_Log2_Hist.pdf | PDF File | |
| JPJMs_Genes_Log2_Hist.pdf | PDF File | |
| JPJMs_Transcripts_Log2_Hist.pdf | PDF File | |
| ExonSkippingEvents_KnownVsNovel_LinePlot.pdf | PDF File | |
| PercentGeneJunctionsCovered_BreadthvsDepth_BoxPlot.pdf | PDF File | |
| IntronSizes_ObservedVsKnown_Density.pdf | PDF File | |
| Stats.tsv | Text File |
| Name | Type | Description |
|---|---|---|
| *.metrics | Symbolic Link | |
| *-AlignmentLengthDistribution.tsv | Text File | |
| *-ErrorRate.tsv | Text File | |
| *-ErrorRate_rates_1.png | Image File | |
| *-ErrorRate_rates_2.png | Image File | |
| *-PicardGC_chart.pdf | PDF File | |
| *-PicardGC_metrics.txt | Text File | |
| *-PicardGC_summary.txt | Text File | |
| *-PicardMetrics.alignment_summary_metrics | Text File | |
| *-PicardMetrics.insert_size_histogram.pdf | PDF File | |
| *-PicardMetrics.insert_size_metrics | Text File | |
| *-PicardMetrics.quality_by_cycle.pdf | PDF File | |
| *-PicardMetrics.quality_by_cycle_metrics | Text File | |
| *-PicardMetrics.quality_distribution.pdf | PDF File | |
| *-PicardMetrics.quality_distribution_metrics | Text File | |
| *-ReadLengthDistribution.tsv | Text File | |
| *-ReadLengthSummary.tsv | Text File | |
| *.bam | Symbolic Link | |
| *.bam.bai | Symbolic Link | |
| *.bam.flagstat | Symbolic Link | |
| *.bam.html | HTML File | |
| *_fastqc | Directory | |
| *_fastqc.zip | Compressed Archive |
| Name | Type | Description |
|---|---|---|
| summary.txt | Text File | |
| fastqc_report.html | HTML File | |
| fastqc_data.txt | Text File | |
| Icons | Directory | |
| Images | Directory |
| Name | Type | Description |
|---|---|---|
| error.png | Image File | |
| fastqc_icon.png | Image File | |
| tick.png | Image File | |
| warning.png | Image File |
| Name | Type | Description |
|---|---|---|
| per_base_quality.png | Image File | |
| per_sequence_quality.png | Image File | |
| per_base_sequence_content.png | Image File | |
| per_base_gc_content.png Image | File | |
| per_sequence_gc_content.png | Image File | |
| per_base_n_content.png | Image File | |
| sequence_length_distribution.png | Image File | |
| duplication_levels.png | Image File |
The logs/ directory contains the standard error and standard out of each cluster job generated during the build process. These log files are useful for debugging failures, but their diversity and format are beyond the scope of this documentation.
Cufflinks has three modes: de novo, reference guided and reference only. This symlinked directory contains the output of Cufflinks run in the 'de novo' mode. All Cufflinks output files are well documented in the Cufflinks user manual: http://cufflinks.cbcb.umd.edu/manual.html#cufflinks_output
Cufflinks has three modes: de novo, reference guided and reference only. This symlinked directory contains the output of Cufflinks run in the 'reference guided' mode. All Cufflinks output files are well documented in the Cufflinks user manual: http://cufflinks.cbcb.umd.edu/manual.html#cufflinks_output
Cufflinks has three modes: de novo, reference guided and reference only. This symlinked directory contains the output of Cufflinks run in the 'reference only' mode. All Cufflinks output files are well documented in the Cufflinks user manual: http://cufflinks.cbcb.umd.edu/manual.html#cufflinks_output
The expression/ symlink points to the 'reference only' Cufflinks mode. This symlink exists simply to maintain backward compatibility with existing software.
The results directory is a base-level directory to serve as a container for all build results. Newer pipelines use this convention and future results will be added to this directory
| Name | Type | Description |
|---|
- | Symbolic Link | A symlink to each instrument data run of htseq-count.
| Name | Type | Description |
|---|---|---|
| transcript-counts.tsv | Text File | |
| transcript.err | Text File | |
| gene-counts.tsv | Text File | |
| gene.err | Text File |
All TopHat files are well documented in the TopHat user manual:
http://tophat.cbcb.umd.edu/manual.shtml#output
All Cufflinks output files are well documented in the Cufflinks user manual:
http://cufflinks.cbcb.umd.edu/manual.html#cufflinks_output
Why did the RNA-seq metrics report a large amount of intronic sequence?
http://www.biostars.org/p/42890/
How do I list the BAM location for a model?
Models do not have results of their own--they are merely a container for builds. Thus we need to select a build from the model. "last_succeeded_build" (and "last_complete_build", which is synonymous) give us the most recent build that completed successfully.
"whole_rmdup_bam_file" is something particular to reference alignment builds. Both reference alignment and rna-seq have a merged_alignment_result (which is itself a Genome::InstrumentData::AlignmentResult::Merged). Looking in that module we see that there is a bam_path accessor.
$ genome model list --filter id=abcd1234 --show id,name,last_complete_build.merged_alignment_result.bam_path
For strand specific library construction protocols, which settings do I use for each RNA-seq software tool? TopHat
http://tophat.cbcb.umd.edu/faq.shtml#library_type
Illumina specifically documents the library type in the Illumina RNA-seq Tophat Analysis (originally downloaded from http://www.illumina.com/documents/products/technotes/RNASeqAnalysisTopHat.pdf): "For the TruSeq RNA Sample Prep Kit, the appropriate library type is 'fr-unstranded'. For TruSeq stranded sample prep kits, the library type is specified as 'fr-firststrand'."
This blog post is also very informative: How to tell which library type to use (fr-firststrand or fr-secondstrand)?
Another suggestion is to load the aligned reads in IGV and look at the read orientation. You can do this by hovering over a read aligned to an exon.
F2 R1 means the second read in the pair aligns to the forward strand and the first read in the pair aligns to the reverse strand. For a positive DNA strand transcript (5' to 3') this would denote a fr-firststrand setting in TopHat, ie. "the right-most end of the fragment (in transcript coordinates) is the first sequenced". For a negative DNA strand transcript (3' to 5') this would denote a fr-secondstrand setting in TopHat.
F1 R2 means the first read in the pair aligns to the forward strand and the second read in the pair aligns to the reverse strand. See above for the complete definitions, but it's simply the inverse for F1 R2 mapping.
Anything other than FR orientation is not covered here and discussion with the individual responsible for library creation would be required. Typically RF orientation is reserved for large-insert mate-pair libraries. Other orientations like FF and RR seem impossible with Illumina sequence technology and suggest structural variation between the sample and reference.
http://www.biostars.org/p/42890/
Models do not have results of their own--they are merely a container for builds. Thus we need to select a build from the model. "last_succeeded_build" (and "last_complete_build", which is synonymous) give us the most recent build that completed successfully.
"whole_rmdup_bam_file" is something particular to reference alignment builds. Both reference alignment and rna-seq have a merged_alignment_result (which is itself a Genome::InstrumentData::AlignmentResult::Merged). Looking in that module we see that there is a bam_path accessor.
$ genome model list --filter id=abcd1234 --show id,name,last_complete_build.merged_alignment_result.bam_path
For strand specific library construction protocols, which settings do I use for each RNA-seq software tool?
http://tophat.cbcb.umd.edu/faq.shtml#library_type
Illumina specifically documents the library type in the Illumina RNA-seq Tophat Analysis (originally downloaded from http://www.illumina.com/documents/products/technotes/RNASeqAnalysisTopHat.pdf): "For the TruSeq RNA Sample Prep Kit, the appropriate library type is 'fr-unstranded'. For TruSeq stranded sample prep kits, the library type is specified as 'fr-firststrand'."
This blog post is also very informative: How to tell which library type to use (fr-firststrand or fr-secondstrand)?
Another suggestion is to load the aligned reads in IGV and look at the read orientation. You can do this by hovering over a read aligned to an exon.
F2 R1 means the second read in the pair aligns to the forward strand and the first read in the pair aligns to the reverse strand. For a positive DNA strand transcript (5' to 3') this would denote a fr-firststrand setting in TopHat, ie. "the right-most end of the fragment (in transcript coordinates) is the first sequenced". For a negative DNA strand transcript (3' to 5') this would denote a fr-secondstrand setting in TopHat.
F1 R2 means the first read in the pair aligns to the forward strand and the second read in the pair aligns to the reverse strand. See above for the complete definitions, but it's simply the inverse for F1 R2 mapping.
Anything other than FR orientation is not covered here and discussion with the individual responsible for library creation would be required. Typically RF orientation is reserved for large-insert mate-pair libraries. Other orientations like FF and RR seem impossible with Illumina sequence technology and suggest structural variation between the sample and reference.
http://picard.sourceforge.net/command-line-overview.shtml#CollectRnaSeqMetrics
In summary, the settings for commonly used RNA-seq library construction kits are:
| Library Kit | 5' to 3' IGV | Tophat | HTSeq | Picard |
|---|---|---|---|---|
| TruSeq | Strand Specific Total RNA | F2R1 | fr-firststrand | reverse |
| NuGEN | Encore | F1R2 | fr-secondstrand | yes |
| NuGEN | OvationV2 | F2R1 or F1R2 | fr-unstranded | no |