Somatic Variation
Processing Profile: 2762562
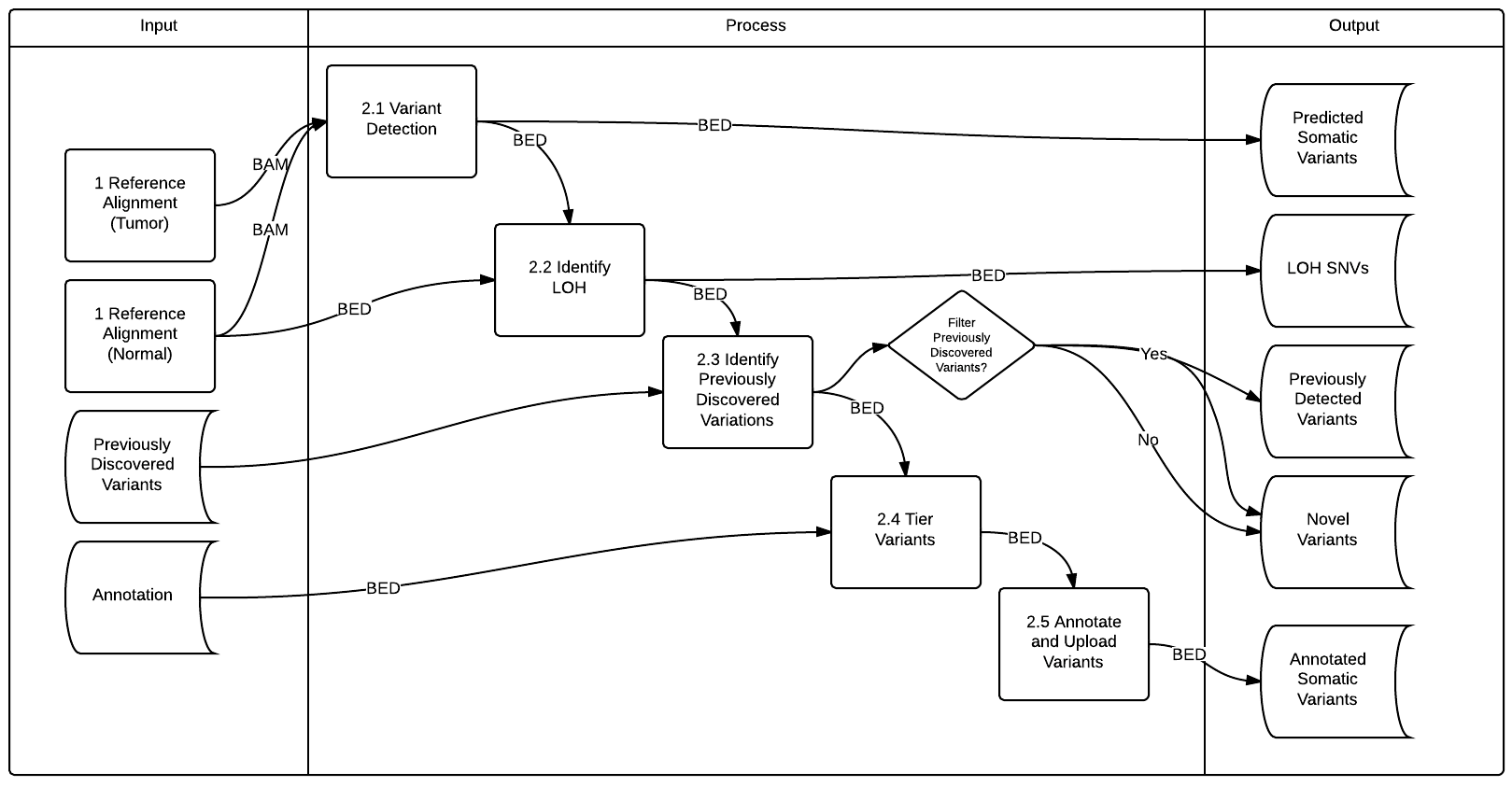
What alignment is done depends on what ReferenceAlignment models were used.
We detected somatic SNVs using Samtools v0.1.1, SomaticSniper2 v1.0.2, Strelka V0.4.6.2, and VarScan v2.2.6
First, Samtools calls were retained if they met all of the following rules inspired by MAQ.
- Site is greater than 10bp from a predicted indel of quality 50 or greater
- The maximum mapping quality at the site is ≥ 40
- Fewer than 3 SNV calls in a 10 bp window around the site
- Site is covered by at least 3 reads and less than 1,000,000,000 reads
- Consensus quality ≥ 20
- SNP quality ≥ 20.
After these filters were applied, Samtools and SomaticSniper calls were unioned using joinx v1.6 (https://github.com/genome/joinx; joinx sort --stable --unique). The resulting merged set of variants were additionally filtered to remove likely false positives2,4. We used bam-readcount v0.4 (https://github.com/genome/bam-readcount) with a minimum base quality of 15 (-b 15) to generate metrics and retained sites based on the following requirements:
- Minimum variant base frequency at the site of 5%
- Percent of reads supporting the variant on the plus strand ≥ 1% and ≤ 99% (variants failing these criteria are filtered only if the reads supporting the reference do not show a similar bias)
- Minimum variant base count of 4
- Variant falls within the middle 90% of the aligned portion of the read
- Maximum difference between the quality sum of mismatching bases in reads supporting the variant and reads supporting the reference of 100
- Maximum mapping quality difference between reads supporting the variant and reads supporting the reference of 30
- Maximum difference in aligned read length between reads supporting the variant base and reads supporting the reference base of 25
- Minimum average distance to the effective 3’ end§ of the read for variant supporting reads of 20% of the sequenced read length
- Maximum length of a flanking homopolymer run of the variant base of 5.
After this filtering, the SomaticSniper/Samtools calls were additionally filtered to high confidence variants by retaining only those sites where:
- The average mapping quality of reads supporting the variant allele was ≥ 40
- The SomaticScore of the call was ≥ 40.
VarScan calls were retained if they met the following criteria:
- VarScan reported a somatic p-value ≤ 0.07
- VarScan reported a normal frequency ≤ 5%
- VarScan reported a tumor frequency ≥ 10%
- VarScan reported ≥ 2 reads supporting the variant.
VarScan variants passing these criteria were then filtered for likely false positives using bam-readcount v0.4 and identical criteria as described above for SomaticSniper. Fully filtered calls as described above for SomaticSniper and VarScan were then merged with calls from Strelka using joinx v1.6 (joinx sort --stable --unique) to generate the final callset.
We detected indels using the GATK 1.0.5336 (-T IndelGenotyperV2 --somatic --window_size 300 -et NO_ET), retaining only those which were called as Somatic, Pindel v0.2.2 (-w 10; with a config file generated to pass both tumor and normal BAM files set to an insert size of 400), Strelka v0.4.6.2 (with default parameters except for setting isSkipDepthFilters = 0), and VarScan v2.2.6 (--min-coverage 3 --min-var-freq 0.08 --p-value 0.10 --somatic-p-value 0.05 --strand-filter 1).
We used bam-readcount v0.4 (https://github.com/genome/bam-readcount) with a minimum base quality of 15 (-b 15) to generate metrics and retained GATK indel sites based on the following requirements:
- Maximum length of an adjacent homopolymer of 4 bases (only affects 1-2 bp indels)
- Percent of reads supporting the indel on the plus strand ≥ 1% and ≤ 99% (indels failing these criteria are filtered only if the reads supporting the reference do not show a similar bias)
- Minimum number of indel supporting reads of 2
- Minimum indel supporting read frequency at the site of 5%
- Indel falls within the middle 90% of the aligned portion of the read
- Maximum difference between the quality sum of mismatching bases in reads supporting the indel and reads supporting the reference of 100
- Maximum mapping quality difference between reads supporting the indel and reads supporting the reference of 30
- Maximum difference in aligned read length between reads supporting the variant base and reads supporting the reference base of 15
- Minimum average distance to the effective 3’ end§ of the read for indel supporting reads of 20% of the sequenced read length.
Pindel calls were retained if they had:
- No support in the normal data
- Had more reads reported by Pindel than reported by Samtools at the indel position or if the number of supporting reads from Pindel was ≥ 8% of the total depth at the position reported by Samtools
- Samtools reported a depth less than 10 at the region and Pindel reported more indel supporting reads than reads mapped with gaps at the site of the call
- A Fisher's exact test p-value ≤ 0.15 was returned when comparing the number of reads with gapped alignments versus reads without in the normal to the tumor
VarScan indel calls were retained if they met the following criteria:
- VarScan reported a somatic p-value ≤ 0.07
- VarScan reported a normal frequency ≤ 5%
- VarScan reported a tumor frequency ≥ 10%
- VarScan reported ≥ 2 reads supporting the variant.
VarScan variants passing these criteria were then filtered for likely false positives using bam-readcount v0.4 and identical criteria as described above for GATK.
Filtered calls from each caller as described above were merged using joinx v1.6 (joinx sort --unique --stable) to generate the final callset.
NA
NA
TBD
TBD
