{kind=link}
This repository contains code to train different convolutional neural networks on deep mutational scanning datasets (or any datasets that associate scores to mutations in a protein) using a new kind of protein structure encoding and to predict the scores for untested variants. Also different methods for data augmentation and pretraining are implemented to get better predictions with less experimental data. It contains supplementary data to the paper "Flattening the curve - how to get better predictions with less data" and code to reproduce these results as well as to train these networks on new datasets.
![]()
Software Requirements:
optional:
File requirements:
- pdb file of the protein of interest
- File containing the proteins structure
- This structure has to have the same amino acid sequence as the mutations used in the deep mutational scanning dataset file
- File Format
- deep mutational scanning dataset as tsv file with 3 tab separated columns:
- sample header: variants
\tnum_mutations\tscore\n - sample row: G1W
\t1\t1.618034\n - variants (describing the variant eg G1W)
- num_mutations (number of mutations introduced by the variant - would be 1 in the example)
- score (the (fitness-) score of the variant
- sample header: variants
- alignment file as clustal without sequence count, a3m or fasta file
- MultipleSequenceAlignment File of the protein
- Must contain the protein of interest's sequence and only unique sequence IDs
- can be omitted but is highly recommended to use
The models can be trained with and without a GPU. However, it is highly recommended to use a GPU.
The models are implemented using tensorflow and keras. An installation guide can be found here.
In order give the program the required settings, one can supply the protein's sequence, the variant column name, the score column name and the number of mutations column name in the deep mutational scanning dataset file as well as the offset (which specifies the index of the first amino acid (starting to count with 0)) using the argparser arguments.
An easier way (especially when done more than once) would be to save it in the datasets/protein_settings_ori.txt file using a unique name in the same format as the examples.
The minimum input to train and save a model would be (assuming basic settings are stored in datasets/protein_settings_ori.txt under PROTEIN_NAME):
python3 d4_cmd_driver.py --protein_name PROTEIN_NAME --pdb_filepath /PATH/TO/PDB/FILE --tsv_filepath /PATH/TO/DEEPMUTATIONALSCANNING/FILE --save_model
Same example without protein settings in protein_settings_ori, but including a MultipleSequenceAlignment:
python3 d4_cmd_driver.py --query_name PROTEIN_NAME_IN_MSA --alignment_file /PATH/TO/ALIGNMENT/FILE --tsv_filepath /PATH/TO/DEEPMUTATIONALSCANNING/FILE --pdb_filepath /PATH/TO/PDB/FILE --number_mutations num_mutations --variants variants --score score --wt_seq PROTEINWTSEQVENCE --first_ind 1
If the proteins pdb file is stored as PROTEIN_NAME.pdb in datasets/ one can omit the --protein_filepath argument if this pdb file should be used.
In order to pretrain the models, a pseudo score for each possible single and double variant gets calculated. This datasets can then be used to pretrain the models on "variants" of the same protein they will later have to predict scores for.
In order to create pretraining data run python3 d4_pt_dataset.py with the needed arguments to create pretraining data.
A minimum setting (if the protein is set in protein_settings_ori.txt) would be python3 d4_pt_dataset.py --p_name PROTEIN_NAME --pdb_file /PATH/TO/PDB/FILE.
PROTEIN_NAME is also important to be the same name as the wild type sequence in the alignment file.
Run it with only the -h flag to see all possible parameters.
After creating the dataset, one can run the same command as in the minimal example above with the pseudo dataset tsv file path used in the --tsv_filepath.
This will save the pretrained model in result_files/saved_models/ containing time and dataset name in its name like dataset_DD_MM_YYYY_HHMM. In there the model with the best validation performance will be stored. The same directory with _end will contain the model in its state at the last training epoch.
All models (no matter if they originate from pretraining or not) will be stored in this way.
After that one can run the training on the real data like:
python3 d4_cmd_driver.py --protein_name PROTEIN_NAME --query_name PROTEIN_NAME_IN_MSA --alignment_file /PATH/TO/ALIGNMENT/FILE --pdb_filepath /PATH/TO/PDB/FILE --tsv_filepath /PATH/TO/DEEPMUTATIONALSCANNING/FILE --transfer_conv_weights /PATH/TO/STORED/MODEL/ --save_model
Where --transfer_conv_weights contains the file path to the directory where the models assets/, keras_metadata.pd, saved_model.pd and variables/are stored (like described above).
By default only the classifier part (fully connected dense layers) and not the feature extraction part (convolution layer) of a "transferred model" are trainable. To train the whole model add --train_conv_layers to the command above.
Since more data usually leads to better results, an data augmentation method, specific to protein score data is implemented. It randomly adds variants and their score to create more data points with the assumption that most of mutations behave (at least kind of) additive.
For instance G1W:1.61 and K42G:4.04 would result in an additional variant G1W,K42G:5.65. This is done multiple times in order to get from 50 to around 4800 data points with a maximum of 20,000 newly created points.
To use that kind of data augmentation, one just needs to add --data_aug. The data will be created each time the program is run with this flag and is not stored. This can be used for pretrained and not pretrained models.
The same as described above can be done via a config.ini file. A sample config file can be found in datasets/config_files/config.ini.
To run the prediction with the config file use:
python3 d4_cmd_driver.py --config_file /PATH/TO/CONFIG/FILE
Some of the most used additional features beside the minimal run arguments described above:
--architecturewhich architecture to used4_models.pycontains a template to create a neural network in a function. This function's name can be passed as the--architectureargument tod4_cmd_driverin order to use the model defined there. This makes it easy to test and train any model architecture on this task.
--training_epochssetting number of training epochs--deploy_early_stopuse early stop so model won't over-fit to much (based ones_patience)--es_patiencepatience (number of epochs the model can try to decrease the validation loss before training stops)--batch_sizebatch size--learning_ratelearning rate--split0,--split1,--split2setting the train/validation/test split sizes (fraction or absolute number)--data_augset to use data augmentation--load_trained_model_pathload any model as long as it accepts input with the correct shape (NxNx7 or NxNx6)--p_dirset absolute path to parent directory of the repository if it's not run from inside the parent directory--jit_compilecan be set to avoid jit compile of tensorflow--write_tempwrites the metrics per epoch toresults/temp.csv
In order to see all possible settings run:
python3 d4_cmd_driver.py -h
The results of the training will be stored in result_files/results.csv. A file with all the used arguments will be stored in result_files/log_file.csv.
Model storage is described in "Creating pretraining dataset(s) above".
A plot of the training history (if --save_fig --validate_training flags are set) will be stored in result_files/plots_dataset_DD_MM_YYYY_HHMM.
Extensive test results (correlation plot, box plot of errors per number of mutations and error histogram/scatter plot - if --save_fig --extensive_test flags are set) will be stored in the same directory as the training history.
In order to predict the score of the variants G1W, K42G and G1W,K42G run:
python3 d4_predict.py --model_filepath /PATH/TO/TRAINED/MODEL/ --protein_pdb /PATH/TO/PDB/FILE --alignment_file /PATH/TO/ALIGNMENT/FILE --query_name PROTEIN_NAME_IN_MSA --wt_seq PROTEINSEQVENCE --variant_s G1W_K42G_G1W,K42G
Or without a MultipleSequenceAlignment:
python3 d4_predict.py --protein_pdb /PATH/TO/PDB/FILE --wt_seq PROTEINSEQVENCE --variant_s G1W_K42G_G1W,K42G --model_filepath /PATH/TO/TRAINED/MODEL/
Different variants need to be _ separated.
- Pretrain models:
bash pretrain.sh- has to be done for each protein and for each architecture
- Train models with different parameter setting (w/ and w/o pretraining and data augmentation):
bash mod_super_loop.sh- the models pretrained models have to be moved in a directory structure like
result_files/saved_models/ARCHITECTURENAME_pretrained_PROTEINNAME/PROTEINNAME_*
- Recall performance:
bash recall_split_train.shbash whole_train.sh- last script has to be done for each protein
- Generalization capability:
bash generalization.sh
@article {Wirnsberger2023.03.27.534314,
author = {Wirnsberger, Gregor and Priti{\v s}anac, Iva and Oberdorfer, Gustav and Gruber, Karl},
title = {Flattening the curve - How to get better results with small deep-mutational-scanning datasets},
year = {2023},
doi = {10.1101/2023.03.27.534314},
}
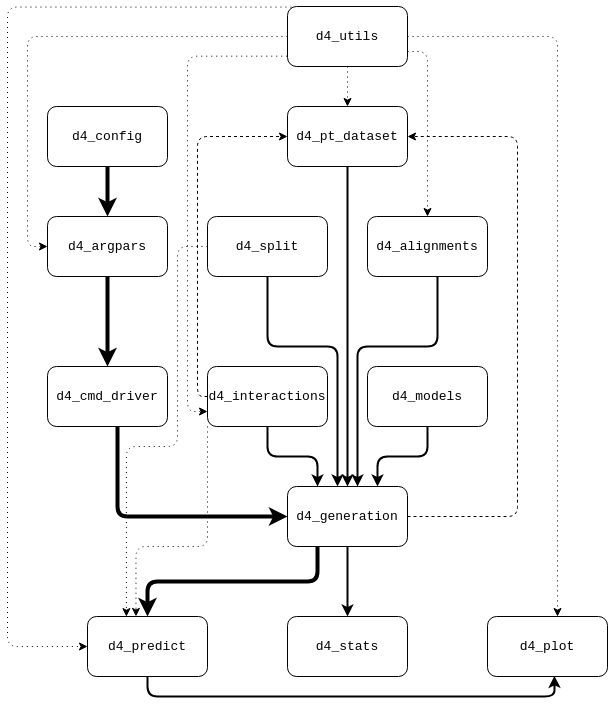
If several directories have the same hierarchy and file content just for different proteins or architectures, only one example is shown
dms
|-- d4_alignments.py
|-- d4_arpars.py
|-- d4_cmd_driver.py
|-- d4_config.py
|-- d4_generation.py
|-- d4_interactions.py
|-- d4_models.py
|-- d4_plot.py
|-- d4_predict.py
|-- d4_pt_dataset.py
|-- d4_split.py
|-- d4_stats.py
|-- d4_utils.py
|-- generalization.sh
|-- mod_super_loop.sh
|-- pretrain.sh
|-- recall_split_train.sh
|-- whole_train.sh
|
|-- datasets/
| |--protein_settings_ori.txt
| |-- alignment_files/
| | |-- avgfp_1000_experimental.clustal
| | |-- gb1_1000_experimental.clustal
| | '-- pab1_1000_experimental.clustal
| |-- config_files/
| | '-- config.ini
| '-- pseudo_scores/
| |-- avgfp/
| | '-- 21x avgfp_*.tsv
| |-- gb1/
| '-- pab1/
|
|-- nononsense/
| |-- first_split_run/
| | |-- avgfp_even_splits/
| | | |-- split_50/
| | | | |-- stest.txt
| | | | |-- train.txt
| | | | '-- tune.txt
| | | |-- split_100/
| | | |-- split_250/
| | | |-- split_500/
| | | |-- split_1000/
| | | |-- split_2000/
| | | '-- split_6000/
| | |-- gb1_even_splits/
| | ’-- pab1_even_splits/
| |-- second_split_run/
| |-- third_split_run/
| |-- single_double_avgfp/
| | '-- avgfp_splits0/
| | |-- stest.txt
| | |-- train.txt
| | '-- tune.txt
| |-- nononsense_avgfp.tsv
| |-- nononsense_gb1.tsv
| |-- nononsense_pab1.tsv
| |-- avgfp_test_formatted.txt
| |-- gb1_test_formatted.txt
| '-- pab1_test_formatted.txt
|
'-- pub_result_files/
|-- rr3
|-- rr5/
| |-- dense_net2/
| | |-- avgfp_log_file.csv
| | |-- avgfp_results.csv
| | |-- gb1_log_file.csv
| | |-- gb1_results.csv
| | |-- pab1_log_file.csv
| | '-- pab1_results.csv
| |-- generalization/
| | |-- log_file.csv
| | '-- results.csv
| |-- recall/
| | |-- fract_splits_log_file.csv
| | |-- fract_splits_results.csv
| | |-- recall_fract_splits/
| | | |-- dense_net2/
| | | | |-- nononsense_avgfp_*_split0/
| | | | |-- nononsense_gb1_*_split0/
| | | | '-- nononsense_pab1_*_split0/
| | | |-- sep_conv_mix/
| | | '-- simple_model_imp/
| | |-- recall_whole_splits/
| | | |-- nononsense_avgfp_*_split0/
| | | |-- nononsense_gb1_*_split0/
| | | ’-- nononsense_pab1_*_split0/
| | |-- whole_log_file.csv
| | '-- whole_results.csv
| |-- sep_conv_mix/
| '-- simple_model_imp/
|
'-- saved_models/
|-- dense_net2_pretrained_avgfp/
| |-- 7x avgfp_fr_*/
| |-- 7x avgfp_sr_*/
| '-- 7x avgfp_tr_*/
|-- dense_net2_pretrained_gb1/
|-- dense_net2_pretrained_pab1/
|-- recall_fract_ds/
| |-- 7x nononsense_avgfp_*/
| |-- 7x nononsense_gb1_*/
| |-- 7x nononsense_pab1_*/
|-- recall_whole_ds/
| |-- 3x nononsense_avgfp_*/
| |-- 3x nononsense_gb1_*/
| |-- 3x nononsense_pab1_*/
|-- sep_conv_mix_pretrained_avgfp/
|-- sep_conv_mix_pretrained_gb1/
|-- sep_conv_mix_pretrained_pab1/
|-- simple_model_imp_pretrained_avgfp/
|-- simple_model_imp_pretrained_gb1/
|-- simple_model_imp_pretrained_pab1/
'-- simple_model_imp_rr3/