

Author: Jiaxuan Wang 📝

🔧 Fix the batch effect on gene expression workflow!
Every Gene Expression study such as RNA-seq or Array, has an question which the researchers want to explain. But in fact, The interested difference often includes experimental operation difference and experimental individual differences. The researcher has to decide how to minimize the impact of technical and other confounding factors on estimation of the environmental or biological factors of interest. We can call it “batch effect”.
Leek et. al (2010) define batch effects as follows: > Batch effects are sub-groups of measurements that have qualitatively different behaviour across conditions and are unrelated to the biological or scientific variables in a study. For example, batch effects may occur if a subset of experiments was run on Monday and another set on Tuesday, if two technicians were responsible for different subsets of the experiments, or if two different lots of reagents,chips or instruments were used.
For remove batch effect. I packages some function which from famous bacth effect adjust package into a new R package fixbatch. Here I outline multiple strategies workflow because I recognizes that there is not a perfect and univers method to adjust batch effects. Maybe we need choose a better method than others, just because it’s result is more predicted for us.
The workflow employs several successive procedures available in :

🇨🇳 流程图中文版本
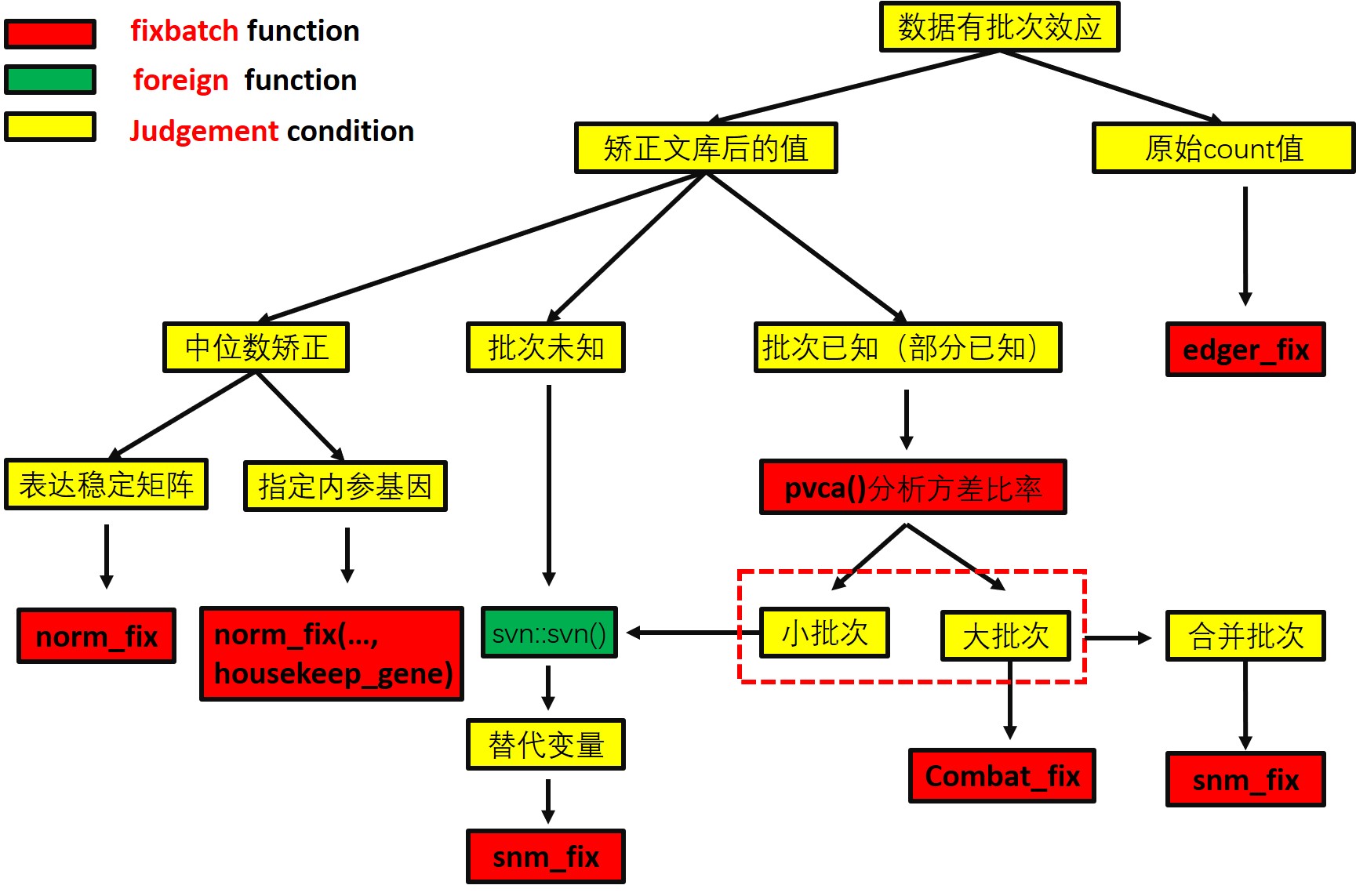{kind=link}
fix bug 2020-12-24
-
add the function to select housekeep gene Separately from every batch group
-
add the function to select some housekeep gene that you know its’ function or famous stable gene like actinB.
-
add the
edger_fixfunction to calculate the adjuts expression cpm value formegdeR
fix bug 2020-12-27
-
add the
pvcafunction form pvca packgas in Bioconductor,this function can help us to estimate factors’ partition of the total variability. I have made some optimizations,such as suppressMessages on useless messages, add test of input data have NAs,fix the the R packages dependency relationship. and addpvca_plotplot function to display the result. -
add the
combat_fixcopy formsva. IT is exactly the same a sva::combat, only changing name. the sva website is “https://bioconductor.org/packages/release/bioc/html/sva.html”
add function 2020-12-29
- add the
snm_fixfunction to fix batch effect and return the normalizeed data. The detailed theory can be found on the website(http://www.bioconductor.org/packages/release/bioc/html/snm.html). I adjusted the input style of parameters to a unified form in fixbatch packages.This is not as simple as I imagined.
the R packages is full fo bugs, beacuse it’s a naive packagse, so I suggest every time when you use the R packages, should update R packages fristly. Remember this step everytimes!
devtools::update_packages("fixbatch")devtools::install_github("wangjiaxuan666/fixbatch")library(fixbatch)calculate the coefficient of variation of the row value in a gene expression matrix.
calculate the sum of rows value in a gene expression matrix.
According to the coefficient of variation and the expression sum, the stable observation value is selected
The function is made for select housekeep gene separately from every batch group
norm_fix allows users to adjust for batch effects in datasets, using the function normalize.quantiles from preprocessCore packages(http://bioconductor.riken.jp/packages/3.6/bioc/html/preprocessCore.html).
The goal of the quantile method is to make the distribution of gene expression intensities for each sample in a set of sample the same. The method is motivated by the idea that a quantile–quantile plot shows that the distribution of two data vectors is the same if the plot is a straight diagonal line and not the same if it is other than a diagonal line. This concept is extended to n dimensions so that if all n data vectors have the same distribution.This suggests we could make a set of data have the same distribution if we project the points of our n dimensional quantile plot onto the diagonal.This implies that we can give each array the same distribution by taking the mean quantile and substituting it as the value of the data item in the original dataset. This motivates the following algorithm for normalizing a set of data vectors by giving them the same distribution.
Calculate normalization factors to scale the raw library sizes. the script is created by Yunshun Chen. The work use the function calcNormFactors and cpm.
I add the combat_fix funtion from svn packages(https://bioconductor.org/packages/release/bioc/html/sva.html). I just rename it, no change for this function. Maybe it will change the form of input parameters later.
The snv package contains functions for removing batch effects and other unwanted variation in high-throughput experiments. Specifically, the sva package contains functions for identifying and building surrogate variables for highdimensional data sets. Surrogate variables are covariates constructed directly from high-dimensional data (like gene expression/RNA sequencing/methylation) that can be used in subsequent analyses to adjust for unknown, unmodeled, or latent sources of noise.
The sva package can be used to remove artifacts in two ways: (1) identifying and estimating surrogate variables for unknown sources of variation in highthroughput experiments and (2) directly removing known batch effects using ComBat.
The function is written based on the ‘pvcaBatchAssess’ function of the PVCA R package(http://watson.nci.nih.gov/bioc_mirror/packages/release/bioc/manuals/pvca/man/pvca.pdf). and adjust by Donghyung Lee for changed slightly to make it more efficient and flexible for sequencing read counts data(https://github.com/dleelab/pvca). But it didn’t update since 4 years ago. My new R packages fixbatch need the function to estimate factor’s partition of the total variability. So I fork the packages and Adjust in my own style.
Principal Variance Component Analysis (PVCA) to explore how technical and biological factors correlate with the major components of variance in the data set; Surrogate Variable Analysis (SVA) to identify major unwanted sources of variation; and Supervised Normalization of Microarrays (SNM) to efficiently remove these sources of variation while retaining the biological factor(s) of interest. The data is based on a study contrasting peripheral blood gene expression in acute myocardial infarction and coronary artery disease patients, described in Kim et al. (2014).
Title A plot function for dispaly the result of principal variance component analysis
The function base on snm R packaegs(http://www.bioconductor.org/packages/release/bioc/html/snm.html), packaged into fixbatch. Citation within R, enter citation(“snm”)).
Leek, J.T. and Storey, J.D. (2007) Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLOS Genetics, 3.
Mecham, B.H., Nelson, P.S. and Storey, J.D. (2010) Supervised normalization of microarrays. Bioinformatics, 26, 1308–1315.
B.M. Bolstad, R.A Irizarry, M. Åstrand, T.P. Speed, A comparison of normalization methods for high density oligonucleotide array data based on variance and bias, Bioinformatics, Volume 19, Issue 2, 22 January 2003, 185–193.