Find_novel_viruses_C_microassembly
[4-8 hours] A high sensitivity search for identifying RNA viruses in the Sequence Read Archive based on Serratus "microassembly".
In brief microassembly is the process of using reads in a library which aligned to the rdrp1 collection for a targetted-assembly of RNA-dependent RNA Polymerase contigs. In contrast to Tutorial B: palmDB Search this does not require an intact palmprint for the search. This has a higher sensitivity for novel viral fragments at the cost of lower specificity/certainty in interpreting divergence values.
- Unix-based command-line
- Query RNA-dependent RNA Polymerase (RdRP) sequences
- Serratus microassemblies:
rdrp1.mu.fa(3.6 GB) -
DIAMOND(v2.0.9+) -
muscle: multiple sequence alignment
The entire RdRP gene is longer than the palmprint sub-sequence used in palmDB, thus the micro-assemblies can contain any sub-sequence of RdRP, missing an intact palmprint (pp negative) of motifs A, B, and C.
virus: -----|========= R D R P =========|-------
motifs: G F A B C D E
palmprint: |======|
pp+ contig: xxxxxxxxxx---------xxxxxxxx (x missing)
pp- contig: -----------------xxxxxxxxxxx
palmprint: |======|
The search queries can contain any RdRP sequences or sub-sequences. InterPro is a great tool for validating for the presence of an RdRP sequence.
For this tutorial we will use a collection of full-length Coronaviridae RdRP sequences coronaviridae.rdrp.fa.
We will be doing a "reverse search": search the contig database against the query since our query is itself a complete collection of CoV RdRP sequences.
# Create diamond database
diamond makedb --in coronaviridae.rdrp.fa -d cov.rdrp
We will use diamond with --ultra-sensitive, -k1 report the single best hit per contig, and -f to explicetly specify output. Since diamond will not report spaces in the output, we modify the rdrp1.mu.fa to change spaces to underscores
# Modify micro-assembly file
sed -i 's/ /_/g' rdrp1.mu.fa
# Run DIAMOND
INPUT='rdrp1.mu.fa'
OUTPUT='rdrp1_cov.out'
DB='cov.rdrp.dmnd'
diamond blastx \
-q $INPUT \
-d $DB \
--masking 0 \
--ultra-sensitive -k1 \
-f 6 qseqid qstart qend qlen \
sseqid sstart send slen \
pident evalue cigar \
qseq_translated \
> $OUTPUT
Which generated the output file: rdrp1_cov.out.
The rdrp1_cov.out file contains 159,742 contigs from 47,247 libraries that potentially match Coronaviridae. This will contain known viruses, novel viruses, and off-target viruses (Nidoviruses). To reduce this list to libraries of interest, we will apply some heuristics. With no semblence of elegance, this will be done in Excel.
- For each library, consider the top hit (highest % identity)
- Matches with >90% identity will be filtered (known)
- Matches with <45% identity will be filtered (non Coronaviridae)
- E-value <1E-6
- Focused on Epsilon-Group Coronaviridae (for tutorial)
This reduces the hits to 120 libraries which will require manual curation and assembly. Note: these microassembly libraries should be treated as hypotheses in which a CoV is potentially present.
The percent identity of the 120 contigs are skewed towards 45% identity, but 68 libraries are still >50%. All of these libraries should undergo complete assembly for validation, but we can spot-check some hits to interpret the results:
Putative Pungitius pungitius Nidovirus (PpungNV).
SRR2976063 is from the Ninespine Stickleback RNA-seq PCBs SLI 9spine_7-1, an isolate from Arctic Ninespine Stickleback (ca. 2016) taken as part of a PCB polluntant study possibly related to von Hippel el at., 2016.
- Serratus.io snapshot shows 29 CoV reads at an average of 66% identity.
DIAMOND output:
contig: SRR2976063_NODE_10_length_227_cov_1.883117
qstart: 67
qend: 216
qlen: 227
rdrp: ERR3994223.Epsy.StypNV
rstart: 285
rend: 334
rlen: 406
id: 76
expect: 1.57E-25
cigar: 50M
qseq: EHMQHTCPLMILSDDGVAGPLTDDPTACCLDDFKATLYYQNNVYLSDEKC
Microassembly:
>SRR2976063_NODE_10_length_227_cov_1.883117
CGCCTATTTCTTTACTCCAGTATGTACGAACATGGTGATTTGGACTCTGCTTCCACGCTA
TTGTTCGAGCATATGCAACACACATGCCCTCTGATGATTCTTTCAGATGACGGCGTTGCT
GGACCTTTAACTGACGACCCAACTGCATGCTGTCTTGATGATTTTAAAGCTACGCTGTAC
TACCAGAACAACGTATATCTTTCTGATGAAAAGTGCTGACCTTGCTA
>SRR2976063_NODE_10_length_227_cov_1.883117_translated
EHMQHTCPLMILSDDGVAGPLTDDPTACCLDDFKATLYYQNNVYLSDEKC
BLASTp Result:
subject: Pacific salmon nidovirus
id: 72.92%
expect: 4e-14
Query 3 MQHTCPLMILSDDGVAGPLTDDPTACCLDDFKATLYYQNNVYLSDEKC 50
M TCPLMILSDDGVAGPL D P A + DF+ATL+YQNNVYL D KC
Sbjct 713 MDDTCPLMILSDDGVAGPLVDHPLAIDVSDFRATLFYQNNVYLDDSKC 760
********
motif C
The microassembly was quite fragmented, yielding 50 aa. This unsurprisingly encompasses motif C, a heavily conserved region which implies the paucity of reads used in the microassembly may be due to limitations in alignment as opposed to low-abundance of viral RNA in the sample. This would be a great candidate for assembly, or even co-assembly with other samples from the same study to recover a complete palmprint, or even a complete genome.
It is worth spot-checking several of the libraries to manually "see" that the search yielded expected results. See: Manual Curation of CoV-Epsy Hits
The 120 libraries (all reads) are fully assembled with coronaSPAdes and the contigs are searched with DIAMOND as in step 3. For the case study this recovers the original contig of 227 nt and a second matching contig of 338 nt (107 aa). Using HMM models for Coronaviridae, additional viral fragments can also be recovered and annotated on a case-by-case basis.
DIAMOND outputs:
contig: SRR2976063_NODE_49267_length_338_cov_1.000000
qstart: 12
qend: 332
qlen: 338
rdrp: SRR6788790.Epsy.AmexNV
rstart: 166
rend: 271
rlen: 411
id: 64.5
expect: 7.26E-43
cigar: 100M1I6M
qseq: KCDRSMPNGLRQAAAWMFLNKHGSCCTHSERTFLLCNEMAQVLNETCVVSGFLYRKPGGTSSGDATTAYANSAFNLFQVVTSALNHLVKRDWPEDTKGNTCRPLVNN
contig: SRR2976063_NODE_105942_length_227_cov_1.883117
qstart: 67
qend: 216
qlen: 227
rdrp: ERR3994223.Epsy.StypNV
rstart: 285
rend: 334
rlen: 406
id: 76
expect: 1.57E-25
cigar: 50M
qseq: EHMQHTCPLMILSDDGVAGPLTDDPTACCLDDFKATLYYQNNVYLSDEKC
Assembly:
>NODE_49267_length_338_cov_1.000000
TCGTAGGTTCAAAATGCGACCGTTCTATGCCTAACGGTTTGAGACAAGCTGCTGCTTGGA
TGTTCTTAAACAAACATGGTTCTTGCTGCACCCACAGTGAACGTACCTTCTTGCTTTGCA
ACGAGATGGCTCAAGTTCTTAATGAAACTTGCGTTGTCAGTGGGTTTCTTTACAGAAAAC
CCGGTGGTACTTCATCTGGTGATGCTACAACAGCTTATGCCAACTCAGCCTTCAACCTAT
TTCAGGTCGTTACAAGCGCACTTAACCATCTCGTTAAGCGTGATTGGCCTGAAGACACAA
AAGGCAACACTTGCCGCCCACTCGTTAACAATCCAACG
BLASTp
## Fragment 1
subject: Pacific salmon nidovirus
id: 70.33% (85% query)
expect: 5e-34
Query 1 KCDRSMPNGLRQAAAWMFLNKHGSCCTHSERTFLLCNEMAQVLNETCVVSGFLYRKPGGT 60
KCDRSMPN LR +AAWMFLNKH CC+ + + L NE++QVL+ETCVV+G YRKPGGT
Sbjct 591 KCDRSMPNLLRMSAAWMFLNKH-PCCSLLDNKYRLANELSQVLSETCVVNGAFYRKPGGT 649
******
motif A (partial)
Query 61 SSGDATTAYANSAFNLFQVVTSALNHLVKRD 91
SSGDATTAYANSAFNLFQV+TSA+N + D
Sbjct 650 SSGDATTAYANSAFNLFQVITSAVNRCMSTD 680
**************
motif B
## Fragment 2 (as above)
Query 3 MQHTCPLMILSDDGVAGPLTDDPTACCLDDFKATLYYQNNVYLSDEKC 50
M TCPLMILSDDGVAGPL D P A + DF+ATL+YQNNVYL D KC
Sbjct 713 MDDTCPLMILSDDGVAGPLVDHPLAIDVSDFRATLFYQNNVYLDDSKC 760
********
motif C
Combined alignment of both fragments to Pacific Salmon Nidovirus with catalytic motifs shown. The similar identity suggests this comes from the same novel coronavirus, but the assembly is incomplete due to low coverage (approx. 1x) although this cannot be known with certainty. The partial motif A and motif B and C are recovered provides a near complete estimate of the palmprint identity of a putative virus rdrp, well within novel species/genera range.
Query 1 KCDRSMPNGLRQAAAWMFLNKHGSCCTHSERTFLLCNEMAQVLNETCVVSGFLYRKPGGT 60
KCDRSMPN LR +AAWMFLNKH CC+ + + L NE++QVL+ETCVV+G YRKPGGT
PsNV 591 KCDRSMPNLLRMSAAWMFLNKH-PCCSLLDNKYRLANELSQVLSETCVVNGAFYRKPGGT 649
******
motif A (partial)
Query 61 SSGDATTAYANSAFNLFQVVTSALNHLVKRDPEDTKGNTCRPLVNN------------EH 107
SSGDATTAYANSAFNLFQV+TSA+N + D
PsNV 650 SSGDATTAYANSAFNLFQVITSAVNRCMSTDRFDYRSFNHELYSNIYLLDTPDRGFVEKF
**************
motif B
Query 3 MQHTCPLMILSDDGVAGPLTDDPTACCLDDFKATLYYQNNVYLSDEKC 50
M TCPLMILSDDGVAGPL D P A + DF+ATL+YQNNVYL D KC
PsNV 709 YNFMDDTCPLMILSDDGVAGPLVDHPLAIDVSDFRATLFYQNNVYLDDSKCWTEADVLKG 769
********
motif C
In the coronaSPAdes output, there are longer non-RdRP fragments detected via HMM models, including a 382 aa methyltransfransferase domain.
# BGC Statistics file
...
BGC subgraph 15
# domains in the component - 3
# Strong/weak edges in the component - 2/0
BGC candidate 1
CoV_Methyltr_2-CoV_NSP15_C-CoV_Methyltr_2
Predicted type: Custom
Domain cordinates:
Edge order:
3282135-
Path is linear
...
>EDGE_3282135_Translated
MNRCSLFMPLPPVLANLSLDTIHKYSTWSLPLQHSLEANLTSDMPTSATLNVLSFDKVV
VGAQSNLVVSAIPLAGKLSRRFARGTLPGCIDGVPTTNPTDTYLTYYVLGKPTDINHVY
FSAGRKVDNFTYYSPIEQDFLTLTPDDFIAKYDLSSFKIKHIIYGEDNKPVVGGLHLLI
SFVLHRHNNTVSFAELTPNDTITVRTAQVSFEASSKVTTFTDLKLDDFICMLKQIDQTT
VSKVMLFNIDFQQIRFMTWNSEGTTLTFYPQPASLQSQSNPVIKVPRSVLFQDLENTKI
LIPNYGKTFKSPYTAGVSICYMKYMQLLHFLDRGTTMAKVGDGKMNILNLGAASIEGSS
PGTDAIKYFFSPLDGIHKQTLQ
BLASTp of the methyltransferase domain shows the weaker conservation in this gene (34% vs 70%). This is as expected as the RDRP is the most highly conserved gene in Coronaviridae (and most RNA viruses).
subject: Pacific salmon nidovirus
id: 34.29% (97% query)
expect: 2e-42
Query 1 MNRCSLFMPLPPVLANLSLDTIHKYSTWSLPLQHSLEANLTSDMPTSATLNVL--SFDKV 58
M RC +FMP L NL ++ I +S +SLP+ +S+ + + D+P + + L +F K+
Sbjct 1997 MYRCCIFMPNNQFLHNLDFNYICGFSNYSLPVSYSVMSGFAGDVPVAMVKSTLGLAFGKI 2056
Query 59 VVGAQSNLVVSAIPLAGKLSRRFARGTLPGCIDGVPTTNPTDTYLTYYVLGKPTDINHVY 118
LV+S P+ S + +G G IDGV N + Y TY + P Y
Sbjct 2057 PFADFMGLVISNDPINST-SVLYHKGETSGIIDGVFALNVINVYYTYVNMPIPPV---TY 2112
Query 119 FSAGRKVDNFTYYSPIEQDFLTLTPDDFIAKYDLSSFKIKHIIYGEDNKPVVGGLHLLIS 178
S R + F + +E D+L+L+ DFI KY L +F ++HI+YGE + P VGGLHL+IS
Sbjct 2113 MSVNRSLLTFIPMTAMEDDYLSLSSTDFITKYSLENFNLEHILYGEFSSPTVGGLHLMIS 2172
Query 179 FVLHRHNNTVSFAELTPNDTITVRTAQVSFEASSKVTTFTDLKLDDFICMLKQIDQTTVS 238
H + +F T + +V + Q+ S+K++T+ D+ +D F+ +LK +D T VS
Sbjct 2173 VFKHYKSYDATFHNRTVSSLSSVLSYQIVTRDSTKLSTLIDITIDSFVAILKSLDLTVVS 2232
Query 239 KVMLFNIDFQQIRFMTWNSEGTTLTFYPQPASLQSQSNPVIKVPRSVLFQDLENTKILIP 298
KV+ ID + I FM W G TFYP P LQ N VIK +V + +I
Sbjct 2233 KVVHIPIDGKPIDFMLWAHAGNVATFYPLPQVLQ---NSVIKRVSAVSIDVALKSSCIIN 2289
Query 299 NYGKTFKSPYTAGVS 313
G+ +P GV+
Sbjct 2290 ASGQFGFTPTCGGVA 2304
This demonstrates the utility of the palmprint sequence quite well. It provides an evolutionary unit measurement within the conserved RdRP from which to infer how related/distant two species are from one another. Since other genes, or even regions of genes diverge at different rates, using the same region of one gene allows for a common method for comparing RNA virus divergence, but as we see here, an intact palmprint is required to do so.
After spot-checking a few datasets we convince ourselves that we're in the correct vicinity for novel viruses. From the complete assembly of these 120 libraries there are 29732 contigs for which DIAMOND returned a potential rdrp match in epsy_120_diamond_cov.out. These sequences were then also searched against the nr database to deplete known sequences.
Sequences are filtered down to 54 putative contigs from 32 librariesi n epsy_120_putative_cov.fa.
- <90% identity to
nrdatabase - <90% and >50% identity match to a CoV RdRP
- <1E-5 expect value
- Length >30 amino acids rdrp-match
cat epsy_120_putative_cov.fa
>ERR3994210_NODE_7_length_7085_cov_12.368249
TVGHYFKDYEAGCLESEYIVVNNPKKSSGFPFSQVGNAGEFLDAMGHERLNQLHKYTRRNILPTFTKVNTKCAISAKDRLRTVAGVGILSTANNRMTHQRMLKSIAAARDKTIVIGTPKFYGNWDSMLSRFNDGVPRELFGWDYPKCDRSMPNMLRMSAAYMFLNKHHDCCDSLTRFYRLTNEMQQVLNETCLVNGTMFRKPGGTSSGDATTAYANSAFNLIQVITSVIDHILCVDWPNDDGCNDNPRRAFANKIYDAIYHSLNVDDVSTTLFDYVQKSCPLMILSDDGVAGRDITDPQSVTLVDFRSTLYYQNNVYLPDNKCWIQPDVTKGPEEFCSQHTILYNGVYLPVPDPSRILAACCYTSTAERADPQLMLERLVSLAIDAYPLVHHSDSTY
>SRR12718429_NODE_966_length_1551_cov_7.324763
EMCQVLTETCLVNGYLYSKPGGTTSGDATTAYANSIFNLFQFITSAVDVFIRRQWKGDNEHVDELQMSNWLYDAMYNGQPAGYRSHIARAFFERMKVKCPLLILSDDGVAGPAIDDPAKLELSDFQNTLFYQNSVFLPANKCWTETNVKVGAHEFCSQHTILHNGIYYPTPDPSRILSACVFSNSTIKADPELMLERLVSLAIDAYPLVFHENNTY
>SRR6233344_NODE_64336_length_499_cov_1.946009
PLSICGNAGAVYKALGHDEIDTLYDYSKRNITPTITKLNAKCALSAKPRVRTVAGVGLVSTMVNRCCYQKKLKSIAQSRGIPIVIGTPKYYGGWNTMVQQVLPPNCKKLFGYDFPKCDRAMPNILRIAADEILLSGHVGNCCSESDVFYRLSNEASQVLHEAVYIE
>SRR9680856_NODE_41808_length_500_cov_2.669704
RTLIEGFDAPDKVLFGWDYPKCDRSMPNILRLVSSLVLSRCHVNCCNNKERFYRLANEAAQVLNEFSYCNGTFYLKPGGTSSGDAITAYANSVFNCCQVATACVTAFLSKNTSTFASLIECKQMQLKLYENIYINSLSYVDKDFVSSYKNFLHQYFNLMILSDDGA
>SRR1324960_NODE_19088_length_569_cov_1.440385
CLHSKCAPFVDFQSQLRSNLYDCVDDPLFVTQYREFLQHTCPLLILSDDGIAGPLADHPLALTLSDFRATLFYQNNVYLSDSKCWVEPDVDNGPEEFCSQHTIVYNGCLYPIPDPSRIIAACCFVSSAEKSDATMQLTRLVSLAIDGYPLIYHKDETF
...
To interpret these contigs, they are sequentially added to a Epsy RdRP multiple sequence alignment.
mkdir msa; cd msa
# Split the epsy file into individual files
seqkit split -i ../epsy_120_putative_cov.fa -O epsy120
cd epsy120
cp ../epsy.rdrp.msa.fa ./msa.in
for file in $(ls *.fa); do
muscle -profile -in1 msa.in -in2 $file -out msa.out
mv msa.out msa.in
done
mv msa.in ../epsy120.putative.msa.fa
Visualizing the MSA we can see the heavy fragmentation within these libraries, yet the sequences show clear homology upon inspection, especially when focusing upon the palmprint motifs A, B and C.
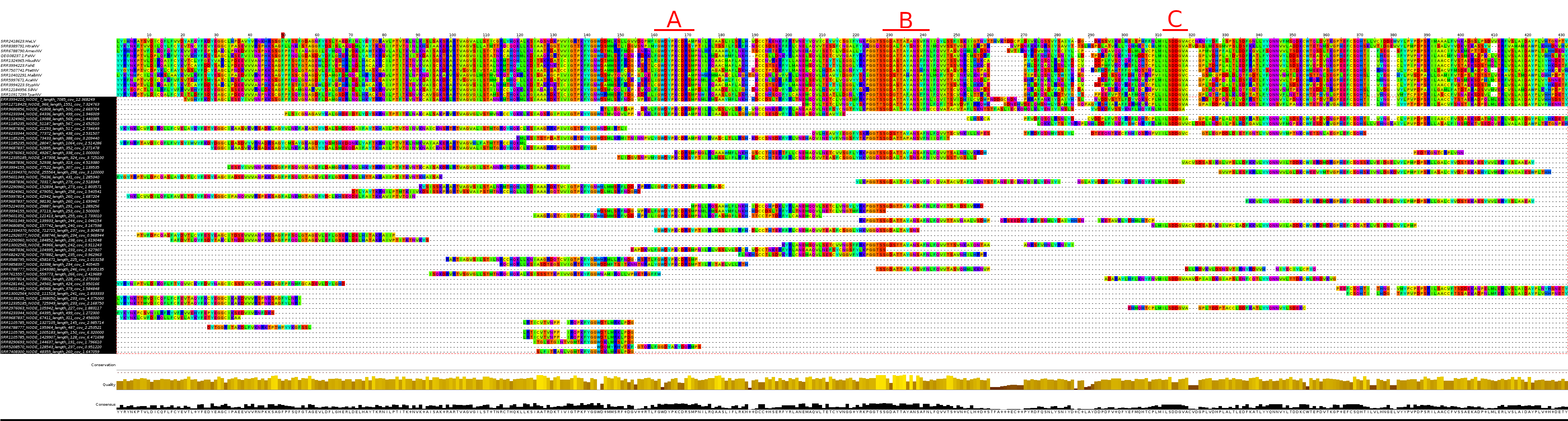

Analyzing the multiple-sequence alignment as well as the origins of these samples, the 53 contigs are estimated to represent an additional 25 distinct novel coronaviruses. It is impossible to make this conclusion without more complete sequences such that each sequence can undergo pair-wise comparison (such as an intact palmprint), but accounting for the duplicate sequences or similar libraries (same sample or organism) supports this. In the very least this provides breadcrumbs for viral epidemiology, if a novel sequence is identified, these fragments can be used to locate similar samples.
That's it! You've used the Serratus microassembly data to mine for additional viruses.
- Coronavirus Microassembly Fragment Annotation
- Tutorial: Running DIAMOND nr search
- Tutorial A: Serratus.io Lookup
- Tutorial B: RdRP palmprint sequence-search